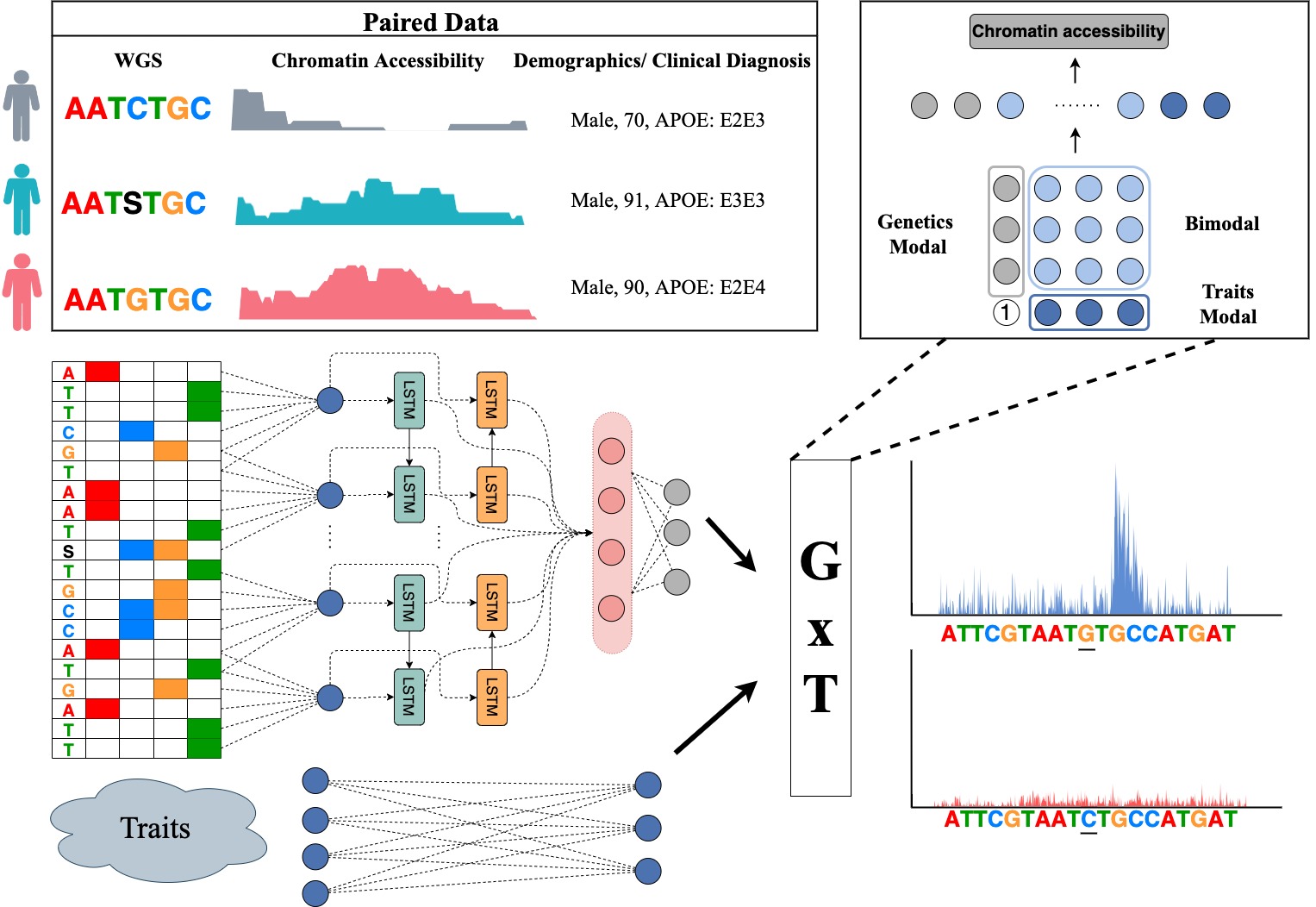
By leveraging paired whole genome sequencing data and epigenetic functional assays in a population cohort, a DeepPerVar is a multi-modal deep learning framework to predict genome-wide quantitative epigenetic signals and evaluate the functional consequence of noncoding variants on an individual level by quantifying their allelic difference on the prediction. By applying DeepPerVar to the ROSMAP cohort studying Alzheimer’s disease (AD), the web server can accurately predict genome-wide H3K9ac signals and DNA methylation ratio given DNA genomic sequence under reference and alternative alleles, and use the allelic difference as the score to evaluate the functional consequence of genetic variants associated with Alzheimer’s disease in a personal genome.
We implement a webserver to predict genome-wide H3K9ac signals and DNA methylation ratio and the mutation effect on these two epigenetics signals. The webserver can be accessed from link.
DeepPerVar is implemented by Python3.
- Python 3.8
- numpy >= 1.18.5
- pytorch ==1.7.1
- biopython=1.19.2
Download Reference Genome (hg19), and put them in the DeepPerVar root directory. Download DeepPerVar Models, and put model files in models directory.
unzip Models.zip Reference.zip
Download DeepPerVar:
git clone https://github.com/alfredyewang/DeepPerVar
Install requirements.
pip3 install -r requirements --user
Install Samtools 1.15.1 follow the (instruction)[http://www.htslib.org/download/] .
You can see the input arguments for DeepPerVar by help option:
usage: DeepPerVar.py [-h] [--prediction] [--epigenomics EPIGENOMICS] [--bed BED] [--model_dir <data_directory>] [--res_dir <data_directory>]
DeepPerVar: a multimodal deep learning framework for functional interpretation of genetic variants in personal genome
optional arguments:
-h, --help show this help message and exit
--prediction Use this option for predict DeepPerVar score
--epigenomics EPIGENOMICS
Epigenetics, can be H3K9 or DNA_methylation
--bed BED The Bed file for predicts epigenetics and mutation effects
--model_dir <data_directory>
The model directory for DeepPerVar
--res_dir <data_directory>
The data directory for save results
DeepPerVar takes UCSC Genome Browser BED file. Each line has 5 tab separated fields. The BED fields are:
- The first column: Chromosome name (hg19).
- The second column: Position of SNPs (hg19).
- The third column: The strand information.
- The fourth column: reference allele.
- The fifth column: alternative allele.
python3 src/DeepPerVar.py --prediction --epigenomics H3K9 --bed data/snps.bed --res_dir res --model_dir models
Results will be save into res/Results_histone.csv
chr pos strand ref alt H3K9AC_REF_Pred H3K9AC_ALT_Pred DELTA_H3K9AC
1 1265154 - T C 18.415241 18.509096 0.093854904
1 1265460 - T A 17.707266 17.64615 -0.061115265
1 2957600 - T C 10.322433 10.464524 0.1420908
1 3691528 - A G 16.85876 16.950903 0.092142105
1 8021919 - C G 82.27526 82.20313 -0.072128296
1 8939842 - G A 42.205887 42.33795 0.13206482
1 10457540 - T C 13.674403 13.556186 -0.11821747
1 11072117 - C T 57.86567 56.590023 -1.2756462
1 11072691 - G A 37.507782 37.999027 0.49124527
1 11083408 - G A 16.937225 15.624798 -1.3124275