-
Notifications
You must be signed in to change notification settings - Fork 4
/
README.Rmd
316 lines (245 loc) · 19.1 KB
/
README.Rmd
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
263
264
265
266
267
268
269
270
271
272
273
274
275
276
277
278
279
280
281
282
283
284
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
---
output:
md_document:
variant: markdown_github
---
<!-- README.md is generated from README.Rmd. Please edit that file -->
```{r, echo = FALSE}
knitr::opts_chunk$set(
collapse = TRUE,
comment = "#>",
fig.path = "README-"
)
```
<img src="man/figures/logo.svg" align="right" height="139" />
<!-- badges: start -->
[](https://github.com/natverse/hemibrainr/actions)
[](https://codecov.io/gh/natverse/hemibrainr?branch=master)
[](https://www.tidyverse.org/lifecycle/#experimental)
<!-- badges: end -->
# hemibrainr
The goal of *hemibrainr* is to provide useful code for preprocessing and analysing data from the [Janelia FlyEM hemibrain](https://www.janelia.org/project-team/flyem) project. It makes use of the [natverse](https://github.com/natverse) R package, [neuprintr](https://github.com/natverse/neuprintr) to get hemibrain data from their connectome analysis and data hosting service [neuprint](https://github.com/connectome-neuprint/neuPrint). The dataset has been described [here]((https://www.biorxiv.org/content/10.1101/2020.01.21.911859v1)). Using this R package in concert with the [natverse](https://github.com/natverse/natverse) ecosystem is highly recommended.
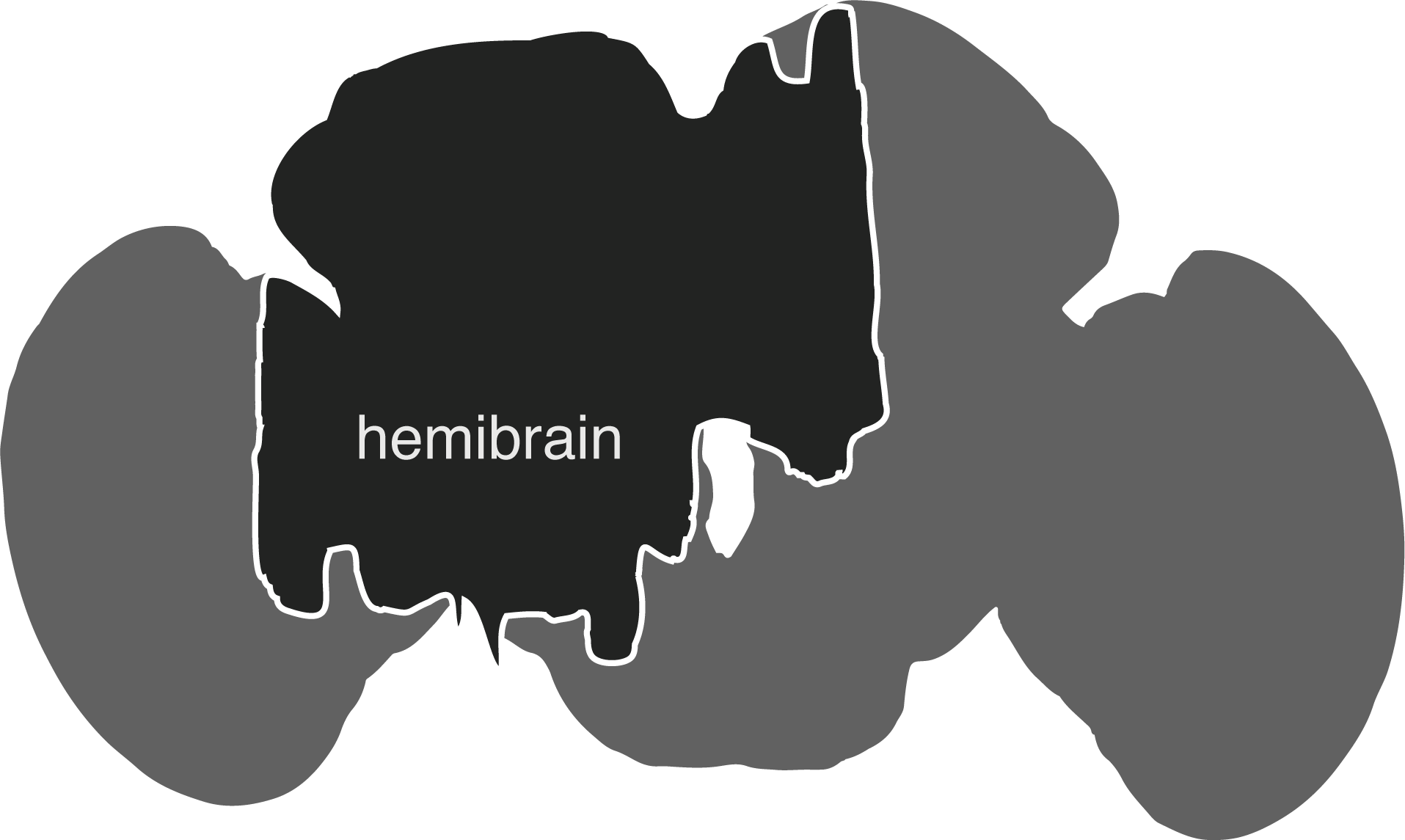
The hemibrain connectome comprises the region of the fly brain depicted below. It is ~21,662 ~full neurons, 9.5 million synapses and is about ~35% complete in this region:
<center>
</center>
## Get started with hemibrainr
### Installation
```{r install, eval = FALSE}
# install
if (!require("remotes")) install.packages("remotes")
remotes::install_github("natverse/hemibrainr")
# use
library(hemibrainr)
```
### Using hemibrainr
*hemibrainr* contains tools with which to quickly work with [hemibrain](https://neuprint.janelia.org/help/videos?dataset=hemibrain) and [FlyWire](https://ngl.flywire.ai/?local_id=c8c06ea181ad5447b04beacfc4cb1b66) neurons, and match up neurons within and between data sets.
If you can connect to the *hemibrainr* google shared drive, this package puts thousands of hemibrain and FlyWire neurons at your fingertips, as well as information on their compartments (e.g. axons versus dendrites), synapses and connectivity and between data set neuron-neuron matches. You can:
* Read thousands of pre-skeletonised FlyWire/hemibrain neurons from Google Drive
* Read FlyWire/hemibrain NBLASTs and NBLASTs to hemibrain neurons
* Read FlyWire/hemibrain neurons that are pre-transformed into a variety of brainspaces
Which is all useful stuff. You can explore our articles for more detailed information on what the package can do, and how to set it up with the data stored on Google drive - but can take a quick tour here:
```{r hemi, eval = FALSE}
# Load package
library(hemibrainr)
# Else, it wants to see it on the mounted team drive, here
options("remote_connectome_data")
# We can load meta data for all neurons in hemibrain
db = hemibrain_neurons()
# And quickly read them from the drive, when we try to plot/analyse them!
hemibrain_view()
plot3d(hemibrain.surf, col = "grey", alpha = 0.1)
plot3d(db[1:10])
```
See which neurons have been matched up:
```{r see.matches, eval = FALSE}
# See matches, you can do this without hemibrain Google Team Drive access
View(hemibrain_matched)
# Get fresh matches, you cannot do this without access
## You will be prompted to log-in through your browser
hemibrain_matched_new <- hemibrain_matches()
## NOTE: includes hemibrain<->FlyWire matches!
```
### neuPrint authentication
In order to use *neuprintr*, which fetches data we want to use with *hemibrainr*, you will need to be able to login to a neuPrint server and be able to access it underlying Neo4j database.
You may need an authenticated accounted, or you may be able to register your `@gmail` address without an authentication process.
Navigate to a neuPrint website, e.g. https://neuprint.janelia.org, and hit 'login'. Sign in using an `@gmail` account.
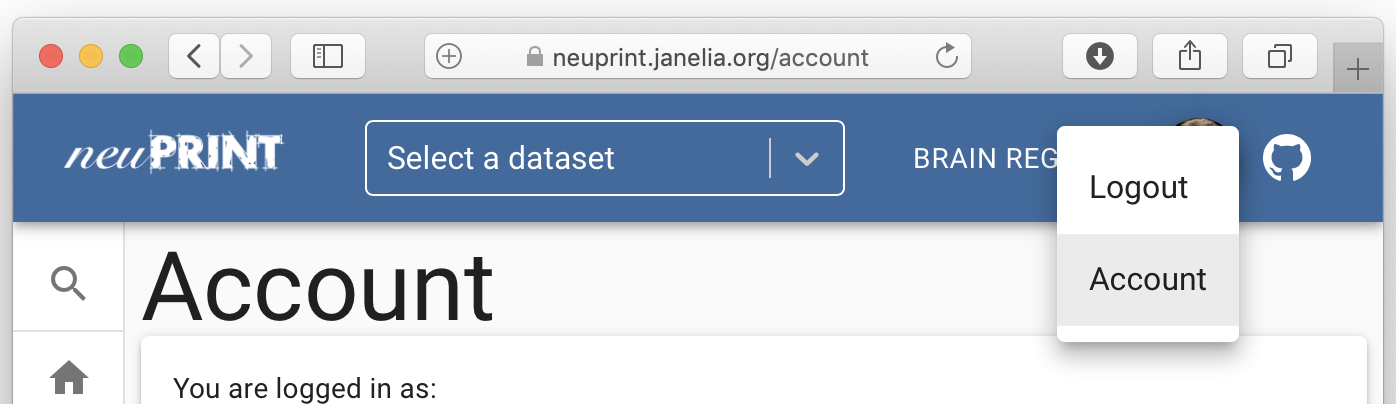
If you have authentication/the server is public, you will now be able to see your access token by going to 'Account':
To make life easier, you can then edit your `.Renviron` file to contain information about the neuPrint server you want to speak with, your token and the dataset hosted by that server, that you want to read. A convenient way to do this is to do
```{r, eval=FALSE}
usethis::edit_r_environ()
```
and then edit the file that pops up, adding a section like
```{r, eval=FALSE}
neuprint_server="https://neuprint.janelia.org"
# nb this token is a dummy
neuprint_token="asBatEsiOIJIUzI1NiIsInR5cCI6IkpXVCJ9.eyJlbWFpbCI6ImIsImxldmVsIjoicmVhZHdyaXRlIiwiaW1hZ2UtdXJsIjoiaHR0cHM7Ly9saDQuZ29vZ2xldXNlcmNvbnRlbnQuY29tLy1QeFVrTFZtbHdmcy9BQUFBQUFBQUFBDD9BQUFBQUFBQUFBQS9BQ0hpM3JleFZMeEI4Nl9FT1asb0dyMnV0QjJBcFJSZlI6MTczMjc1MjU2HH0.jhh1nMDBPl5A1HYKcszXM518NZeAhZG9jKy3hzVOWEU"
```
Make sure you have a blank line at the end of your `.Renviron` file.
For further information try about neuprintr login, see the help for
`neuprint_login()`.
Finally you can also login on the command line once per session, like so:
```{r login2, eval = FALSE}
conn = neuprintr::neuprint_login(server= "https://neuprint.janelia.org/",
token= "asBatEsiOIJIUzI1NiIsInR5cCI6IkpXVCJ9.eyJlbWFpbCI6ImIsImxldmVsIjoicmVhZHdyaXRlIiwiaW1hZ2UtdXJsIjoiaHR0cHM7Ly9saDQuZ29vZ2xldXNlcmNvbnRlbnQuY29tLy1QeFVrTFZtbHdmcy9BQUFBQUFBQUFBDD9BQUFBQUFBQUFBQS9BQ0hpM3JleFZMeEI4Nl9FT1asb0dyMnV0QjJBcFJSZlI6MTczMjc1MjU2HH0.jhh1nMDBPl5A1HYKcszXM518NZeAhZG9jKy3hzVOWEU")
```
This is also the approach that you would take if you were working with more than
two neuPrint servers.
### Connect to hemibrainr Google team drive
For this, you need access to th hemibrainr google team drive. Authentication is through an email account. Once you have access, there are two basic ways to mount the data for use:
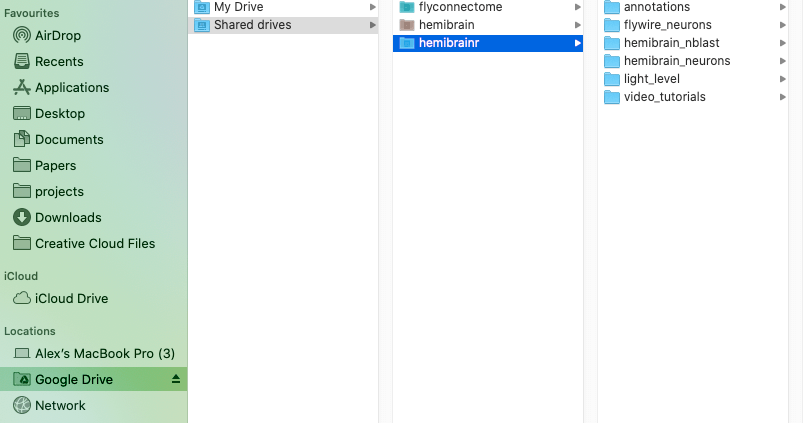
Option 1, mount your Google drives using [Google filestream](https://support.google.com/a/answer/7491144?hl=en). However, for this to work you will need [Google Workspace](https://workspace.google.com/pricing.html), Google's monthly subscription offering for businesses and organizations. One the [Google filestream](https://support.google.com/a/answer/7491144?hl=en) application is run, you should be able to see your drives mounted like external hard drive, as so:
<center>
</center>
Then, this should work:
```{r gdrive, eval = FALSE}
# Set a new Google drive, can be the team drive name or a path to the correct drive
hemibrainr_set_drive("hemibrainr") # No need to run this each time though, this is the default. Use if you want to use a different name drive.
# Now just get the name of your default team drive.
## This will be used to locate your team drive using the R package googledrive
hemibrainr_team_drive()
```
Option 2, this is free. You still need authenticated access to the hemibrainr Gogle team drive. It can then be mounted using [rclone](https://rclone.org/).
First, [download](https://rclone.org/downloads/) rclone for your operating system. You can also download from your system's command line (e.g. from terminal) and then configure it for the drive:
```{r rclone_download, engine = 'bash', eval = FALSE}
# unix/macosx
curl https://rclone.org/install.sh | sudo bash
rclone config
```
And now check this has worked:
```{r rclone_hemibrainr_mount, eval = FALSE}
# mounts in working directory
hemibrainr_rclone()
# Now hemibrain neurons are read from this mount
db = hemibrain_neurons() # read from the google drive
length(db)
plot3d(hemibrain_neurons[1:10])
# Specifically, from here
options("remote_connectome_data")
# unmounts
hemibrainr_rclone_unmount()
# And now we are back to:
options("remote_connectome_data")
```
For more detailed instructions, see [this article](https://natverse.github.io/hemibrainr/articles/google_filestream.html).
### Example: 'splitting' neurons
Let's get started with a useful function for splitting a neuron into its
axon and dendrite:
```{r example, eval = FALSE}
# Choose neurons
## These neurons are some 'tough' examples from the hemibrain:v1.0.1
### They will split differently depending on the parameters you use.
tough = c("5813056323", "579912201", "5813015982", "973765182", "885788485",
"915451074", "5813032740", "1006854683", "5813013913", "5813020138",
"853726809", "916828438", "5813078494", "420956527", "486116439",
"573329873", "5813010494", "5813040095", "514396940", "665747387",
"793702856", "451644891", "482002701", "391631218", "390948259",
"390948580", "452677169", "511262901", "422311625", "451987038"
)
# Get neurons
neurons = neuprint_read_neurons(tough)
# Now make sure the neurons have a soma marked
## Some hemibrain neurons do not, as the soma was chopped off
neurons.checked = hemibrain_skeleton_check(neurons, meshes = hemibrain.rois)
# Split neuron
## These are the recommended parameters for hemibrain neurons
neurons.flow = flow_centrality(neurons.checked, polypre = TRUE,
mode = "centrifugal",
split = "distance")
# Plot the split to check it
nat::nopen3d()
nlscan_split(neurons.flow, WithConnectors = TRUE)
```
## Data fields
Here are some of the most useful column entries across these data. Please let me know if I have missed something important:
| column | description | data entity |
| ----------- | ----------- | ----------- |
| flywire_xyz | A cardinal point in raw FlyWire voxel space that defines a neuron | many |
| flywire_id | The unique 16 digit ID for the flywire neuron, changes when neuron edited | many |
| flywire_svid | The supervoxel ID that corresponds to flywire_xyz | flywire_meta |
| cell_type | If neuron matched to a hemibrain neuron we get its cell_type | flywire_meta |
| side | The hemisphere of the brain that the neuron's soma, or else root-point, is thought to be on | flywire_meta |
| status | A manually assigned 'status' from a Drosophila Connectomics Group tracer, indicating the quality of the neuron | flywire_meta |
| skid | The neuron's skeleton ID in CATMAID, if it exists. The CATMAID neuron may have manualyl annotated synapses | flywire_meta |
| fafb_xyz | A cardinal point in FAFB14 voxel space that defines a neuron | flywire_meta |
| ito_lee_hemilineage | A meaningful biological category, the developmental hemilineage to which we think the neuron belongs, in the nomenclature of Ito et al. 2013 | flywire_meta |
| ito_lee_lineage | A meaningful biological category, the developmental lineage (comprises multiple hemilineages) to which we think the neuron belongs, in the nomenclature of Ito et al. 2013 | flywire_meta |
| hartenstein_hemilineage | A meaningful biological category, the developmental hemilineage to which we think the neuron belongs, in the nomenclature of Lovick/Wong et al. 2013, can be matched to similar names in the larval animal | flywire_meta |
| hartenstein_lineage | A meaningful biological category, the developmental (comprises multiple hemilineages) to which we think the neuron belongs, in the nomenclature of Lovick/Wong et al. 2013, can be matched to similar names in the larval animal | flywire_meta |
| gsheet | A Drosophila Connectomics Group googlesheet on which the flywire_id ID is recorded | flywire_meta |
| hemibrain_match | A semi-manually matched hemibrain neuron's bodyID | flywire_meta |
| hemibrain_match_quality | A manually assigned quality for the hemibrain_match | flywire_meta |
| fafb_hemisphere_match | A cardinal point for a neuron on the other hemisphere that has semi-manually been found to match the given neuron | flywire_meta |
| fafb_hemisphere_match.quality | A manually assigned quality for the fafb_hemisphere_match | flywire_meta |
| dataset | The dataset to which the given neuron belongs | hemibrain_matches |
| total_outputs | The total number of output links / postsynapses from the neuron | flywire_meta |
| axon_outputs | The total number of axonal output links / postsynapses from the neuron | flywire_meta |
| dend_outputs | The total number of dendritic output links / postsynapses from the neuron | flywire_meta |
| total_outputs_density | The total number of output links / postsynapses from the neuron, per micron of cable | flywire_meta |
| axon_outputs_density | The total number of axonal links / postsynapses from the neuron, per micron of cable | flywire_meta |
| dend_outputs_density | The total number of dendritic links / postsynapses from the neuron, per micron of cable | flywire_meta |
| total_outputs | The total number of input links / postsynapses from the neuron | flywire_meta |
| axon_inputs | The total number of axonal input links / postsynapses from the neuron | flywire_meta |
| dend_inputs | The total number of dendritic input links / postsynapses from the neuron | flywire_meta |
| total_inputs_density | The total number of input links / postsynapses from the neuron, per micron of cable | flywire_meta |
| axon_inputs_density | The total number of axonal links / postsynapses from the neuron, per micron of cable | flywire_meta |
| dend_inputs_density | The total number of dendritic links / postsynapses from the neuron, per micron of cable | flywire_meta |
| total_length | The total cable_length, in microns, for the neuron A cardinal point in raw FlyWire voxel space that defines a neuro | flywire_meta |
| axon_length | The axonal cable_length, in microns, for the neuron A cardinal point in raw FlyWire voxel space that defines a neuro | flywire_meta |
| dend_length | The dendritic cable_length, in microns, for the neuron A cardinal point in raw FlyWire voxel space that defines a neuro | flywire_meta |
| pd_length | The primary dendrite (linker) cable_length, in microns, for the neuron A cardinal point in raw FlyWire voxel space that defines a neuro | flywire_meta |
| pnt.length | The primary neurite (cell body fibre) cable_length, in microns, for the neuron A cardinal point in raw FlyWire voxel space that defines a neuro | flywire_meta |
| segregation_index | An entropy score for how segregated the neuron's synapses are into axon and dendrite, see Schneider-Mizell et al 2016, eLife | flywire_meta |
| root | The treenode ID (position in .swc file) of the neuron's root | flywire_meta |
| nodes | The number of nodes in the neuron | flywire_meta |
| segments | The number of segments in the neuron | flywire_meta |
| banchpoints | The number of banchpoints in the neuron | flywire_meta |
| endpoints | The number of endpoints in the neuron | flywire_meta |
| n_trees | The number of trees in the neuron, should be 1 | flywire_meta |
| connectors | The number of synapses in the neuron | flywire_meta |
| postsynapse_side_index | Side side of the neuron that receives the most input, calculated as: (no. postsynaptic links on right - no. on left)/total | flywire_meta |
| presynapse_side_index | Side side of the neuron that receives the most output, calculated as: (no. presynaptic links on right - no. on left)/total | flywire_meta |
| axon_postsynapse_side_index | Same as postsynapse_side_index, but only for the axon | flywire_meta |
| axon_presynapse_side_index | Same as presynapse_side_index, but only for the axon | flywire_meta |
| dendrite_postsynapse_side_index | Same as postsynapse_side_index, but only for the dendrite | flywire_meta |
| dendrite_presynapse_side_index | Same as presynapse_side_index, but only for the dendrite | flywire_meta |
| top_nt | The most prominent predicted transmitter for the neurons' presynpses (the modal across all presynaptic links for: the best nt prediction for each synapses above cleft_score of 50) | flywire_meta |
| offset | The index for the Buhmann synapse in the original .sql table | connectors |
| x,y,z | The position of the connection in FlyWire space| connectors |
| scores | The Buhmamnn prediction score for the synapse, unsure definition | connectors |
| top_p | The probability of the top transmitter prediction for the presynaptic end of this connection | connectors |
| top_nt | The top transmitter prediction for the presynaptic end of this connection | connectors |
| gaba | The synister prediction score for gaba | connectors |
| glutamate | The synister prediction score for glutamate | connectors |
| acetylcholine | The synister prediction score for acetylcholine | connectors |
| octopamine | The synister prediction score for octopamine | connectors |
| serotonin | The synister prediction score for serotonin | connectors |
| dopamine | The synister prediction score for dopamine | connectors |
| prepost | Whether the synapse is pre- (0, i.e. output synapse) orr post (1, i.e. input) | connectors |
| segmentid_pre | The segment (?) for the presynaptic side of the link | connectors |
| segmentid_pre | The segment (?) for the postsynaptic side of the link | connectors |
| pre_svid | The flywire supervoxel ID for the presynaptic side of the link | connectors |
| post_svid | The flywire supervoxel ID for the postsynaptic side of the link | connectors |
| pre_id | The flywire_id for the presynaptic side of the link | connectors |
| post_id | The flywire_id for the postsynaptic side of the link | connectors |
| treenode_id | The treenode in the corresponding swc/d to which this synapse is best attached | connectors |
| strahler_order | The strahler order of the branch on which the synapse is positioned | connectors |
| Label | The comparment of the neuron on which the synapse is positioned, 2 = axon, 3 - dendrite, 4 = primary.dendrite, 7 = primary.neurite, 1 = soma | connectors |
| PointNo | The ID for each point in the neuron | swc/d |
| X,Y,Z | The FlyWire coordinate of the point | swc/d |
| W | The width of the point, generally not used | swc/d |
| parent | The Parent node, -1 means root | swc/d |
| post | The number of postsynapses attachec to node | swc/d |
| pre | The Pnumberf of presynapses attached to node| swc/d |
| flow.cent | The synaptic flow at this node, see Schneider-Mizell et al. 2016 | swc/d |
| strahler_order | The strahler order of this node | swc/d |
## Data
* HemiBrain (hemibrain:v1.0) : from ["A Connectome of the Adult Drosophila Central Brain"](https://www.biorxiv.org/content/10.1101/2020.01.21.911859v1) (Xu, et al. 2020)
## Acknowledging the tools
neuPrint comprises a set of tools for loading and analyzing connectome data into a Neo4j database. Analyze and explore connectome data stored in Neo4j using the neuPrint ecosystem: [neuPrintHTTP](https://github.com/connectome-neuprint/neuPrintHTTP), [neuPrintExplorer](https://github.com/connectome-neuprint/neuPrintExplorer), [Python API](https://github.com/connectome-neuprint/neuprint-python).
This package was created by [Alexander Shakeel Bates](https://scholar.google.com/citations?user=BOVTiXIAAAAJ&hl=en) and [Gregory Jefferis](https://en.wikipedia.org/wiki/Gregory_Jefferis). You can cite this package as:
```{r citation, eval = FALSE}
citation(package = "hemibrainr")
```
**Bates AS, Jefferis GSXE** (2020). *hemibrainr: Code for working with data from Janelia FlyEM's hemibrain project.* **R package** version 0.1.0. https://github.com/natverse/hemibrainr