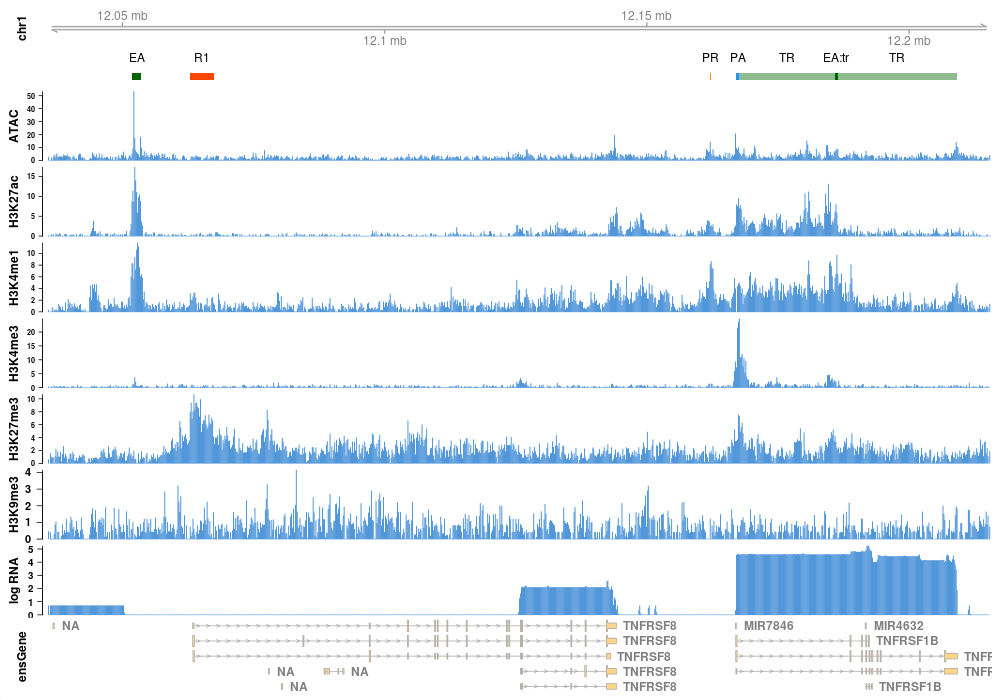
ModHMM is a genome segmentation method that is easy to apply and requires no manual interpretation of hidden states. It provides highly accurate annotations of chromatin states including active promoters and enhancers. The following is a list of states detected by ModHMM:
| State name | Description |
|---|---|
| PA | active promoter |
| EA | active enhancer |
| BI | bivalent region |
| PR | primed region |
| EA:tr | active enhancer in a transcribed region |
| BI:tr | bivalet region in a transcribed region |
| PR:tr | primed region in a transcribed region |
| TR | transcribed region |
| R1 | H3K27me3 repressed |
| R2 | H3K9me3 repressed |
| NS | no signal |
| CL | control signal |
References:
Philipp Benner and Martin Vingron. ModHMM: A Modular Supra-Bayesian Genome Segmentation Method. Journal of Computational Biology. Apr 2020. 442-457. [Link]
E-Mail: <philipp.benner@bäm.de> (replace ä with a).
ModHMM segmentations are available for several ENCODE data sets:
| Tissue | Version | Segmentation | Posteriors | Config | Comment |
|---|---|---|---|---|---|
| GRCh38 ascending aorta | 1.2.2 | Segmentation | Posteriors | Config | total RNA-seq |
| GRCh38 gastrocnemius medialis | 1.2.2 | Segmentation | Posteriors | Config | total RNA-seq |
| GRCh38 heart left ventricle | 1.2.2 | Segmentation | Posteriors | Config | total RNA-seq |
| GRCh38 lung upper lobe | 1.2.2 | Segmentation | Posteriors | Config | total RNA-seq |
| GRCh38 pancreas body | 1.2.2 | Segmentation | Posteriors | Config | total RNA-seq |
| GRCh38 spleen | 1.2.2 | Segmentation | Posteriors | Config | total RNA-seq |
| GRCh38 stomach | 1.2.2 | Segmentation | Posteriors | Config | total RNA-seq |
| GRCh38 tibial nerve | 1.2.2 | Segmentation | Posteriors | Config | total RNA-seq |
| GRCh38 transverse colon | 1.2.2 | Segmentation | Posteriors | Config | total RNA-seq |
| GRCh38 uterus | 1.2.2 | Segmentation | Posteriors | Config | total RNA-seq |
| mm10 forebrain embryo day11.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 forebrain embryo day12.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 forebrain embryo day13.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 forebrain embryo day14.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 forebrain embryo day15.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 forebrain embryo day16.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 heart embryo day14.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 heart embryo day15.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 hindbrain embryo day11.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 hindbrain embryo day12.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 hindbrain embryo day13.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 hindbrain embryo day14.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 hindbrain embryo day15.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 hindbrain embryo day16.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 kidney embryo day14.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 kidney embryo day15.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 kidney embryo day16.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 limb embryo day14.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 limb embryo day15.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 liver embryo day11.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 liver embryo day12.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 liver embryo day13.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 liver embryo day14.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 liver embryo day15.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 liver embryo day16.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 lung embryo day14.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 lung embryo day15.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 lung embryo day16.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 midbrain embryo day11.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 midbrain embryo day12.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 midbrain embryo day13.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 midbrain embryo day14.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 midbrain embryo day15.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
| mm10 midbrain embryo day16.5 | 1.2.2 | Segmentation | Posteriors | Config | poly-A RNA-seq |
ModHMM can be installed by either downloading a binary from the binary repository or by compiling the program from source.
To compile ModHMM you must first install the Go compiler. Afterwards, ModHMM can be installed as follows:
go install -v github.com/pbenner/modhmm@latestAlternatively, it is also possible to clone the repository and compile modhmm manually
git clone github.com/pbenner/modhmm
make
make installUnlike most other genome segmentation methods, ModHMM so far depends on data from a fixed set of features or assays (ATAC/DNase, H3K27ac, H3K9me3, H3K27me3, H3K4me1, H3K4me3, WCE/IgG, and RNA-seq). Open chromatin information must be either provided as ATAC-seq or DNase-seq data. Preferentially, the data should be given as BAM files, but it is also possible to use bigWig files as input.
ModHMM requires a configuration file in JSON format. The following is a simple example where data is provided as BAM files:
{
# Directory containing feature alignment files
"Bam Directory" : ".bam",
# Names of alignment files (for each feature a comma separated list of
# replicates must be specified, any number of replicates is supported)
"Bam Files" : {
"ATAC" : [ "atac-rep1.bam", "atac-rep2.bam"],
#"DNase" : [ "dnase-rep1.bam", "dnase-rep2.bam"],
"H3K27ac" : [ "h3k27ac-rep1.bam", "h3k27ac-rep2.bam"],
"H3K27me3" : ["h3k27me3-rep1.bam", "h3k27me3-rep2.bam"], # optional feature
"H3K9me3" : [ "h3k9me3-rep1.bam", "h3k9me3-rep2.bam"], # optional feature
"H3K4me1" : [ "h3k4me1-rep1.bam", "h3k4me1-rep2.bam"],
"H3K4me3" : [ "h3k4me3-rep1.bam", "h3k4me3-rep2.bam"],
"RNA" : [ "rna-rep1.bam", "rna-rep2.bam"],
"Control" : [ "control-rep1.bam", "control-rep2.bam"] # optional feature
},
# Number of threads used for computing coverage bigWigs (memory intense!)
"Coverage Threads" : 5,
# Directory containing all auxiliary files and the final segmentation
"Directory" : "mm10-liver-embryo-day12.5",
"Description" : "liver embryo day12.5",
# Number of threads used for evaluating classifiers and computing the segmentation
"Threads" : 20,
# Verbose level (0: no output, 1: low, 2: high)
"Verbose" : 1
}ModHMM computes segmentations in several stages. At every stage the output is saved as a bigWig file, which can be inspected in a genome browser. The location and name of each bigWig file can be configured. A full set of all options is printed with modhmm --genconf.
To execute ModHMM simply run (assuming the configuration file is named config.json):
modhmm -c config.json segmentationGenome segmentations are discretized predictions of chromatin states. They do not contain any information about the certainty of a particular prediction. Another drawback is that the number of predicted promoters and enhancers depends on the quality of the data, in particular the sequencing depth. Especially for differential analysis the dependency on the data quality might be hindering. In addition to genome segmentations, ModHMM can compute chromatin state probabilities:
modhmm -c config.json eval-posterior-marginalsThis command will compute genome-wide probabilities for all chromatin states and export them as bigWig files named posterior-marginal-STATE.bw. By default, probabilities are on log-scale. With the option --std-scale ModHMM exports probabilities on standard scale. In this case, bigWig files are named posterior-marginal-exp-STATE.bw.
ModHMM also implements a simple peak-finding algorithm that can be used to call high-probability regions in chromatin state probability tracks:
modhmm -c config.json call-posterior-marginal-peaks --threshold=0.8This command outputs tables with identified peaks, i.e. all regions with probabilities higher than the given threshold.
Most peak callers use a single pre-defined model for computing enrichment probabilities and detecting peaks. In most cases there is a strong model misfit, because of the strong heterogeneity of ChIP-seq data. ModHMM instead allows to fit a mixture distribution (single-feature model) to the observed coverage values with a user-defined set of components. The following command calls ATAC-seq peaks using the estimated single-feature model, if available (see previous section):
modhmm -c config.json call-single-feature-peaks atacThe following configuration can be used if data instead is given in bigWig format:
{
# Data is provided as bigWig files. Set all coverage files static!
"Coverage Files": {
"ATAC" : {"Filename": "coverage-atac.bw", "Static": true },
#"DNase" : {"Filename": "coverage-dnase.bw", "Static": true },
"H3K27ac": {"Filename": "coverage-h3k27ac.bw", "Static": true },
"H3K4me1": {"Filename": "coverage-h3k4me1.bw", "Static": true },
"H3K4me3": {"Filename": "coverage-h3k4me3.bw", "Static": true },
"RNA" : {"Filename": "coverage-rna.bw", "Static": true },
"Control": {"Filename": "coverage-control.bw", "Static": true }
},
# Directory containing all auxiliary files and the final segmentation
"Directory" : "mm10-liver-embryo-day12.5",
"Description" : "liver embryo day12.5",
# Number of threads used for evaluating classifiers and computing the segmentation
"Threads" : 20,
# Verbose level (0: no output, 1: low, 2: high)
"Verbose" : 1
}Coverage bigWig files should contain discrete count data and must be placed in the directory mm10-liver-embryo-day12.5. Setting the option Static to true tells ModHMM that the provided bigWig files are not automatically generated and should not be overwritten.
Create a configuration file named mm10-liver-embryo-day12.5.json (ModHMM accepts an extended JSON format that allows comments):
{
"Bam Directory" : ".bam",
"Bam Files" : {
"ATAC" : ["ENCFF929LOH.bam", "ENCFF848NLJ.bam"],
"H3K27ac" : ["ENCFF524ZFV.bam", "ENCFF322QGS.bam"],
"H3K27me3" : ["ENCFF811DWT.bam", "ENCFF171KAM.bam"],
"H3K9me3" : ["ENCFF293UCG.bam", "ENCFF777XFH.bam"],
"H3K4me1" : ["ENCFF788JMC.bam", "ENCFF340ACH.bam"],
"H3K4me3" : ["ENCFF211WGC.bam", "ENCFF587PZE.bam"],
"RNA" : ["ENCFF405LEY.bam", "ENCFF627PCS.bam"],
"Control" : ["ENCFF865QGZ.bam", "ENCFF438RYK.bam"]
},
"Coverage Threads" : 5,
"Directory" : "mm10-liver-embryo-day12.5",
"Description" : "liver embryo day12.5",
"Threads" : 20,
"Verbose" : 1
}ENCODE bam files will be automatically downloaded by ModHMM.
Create output directory
mkdir mm10-liver-embryo-day12.5Execute ModHMM:
modhmm -c mm10-liver-embryo-day12.5.json segmentation