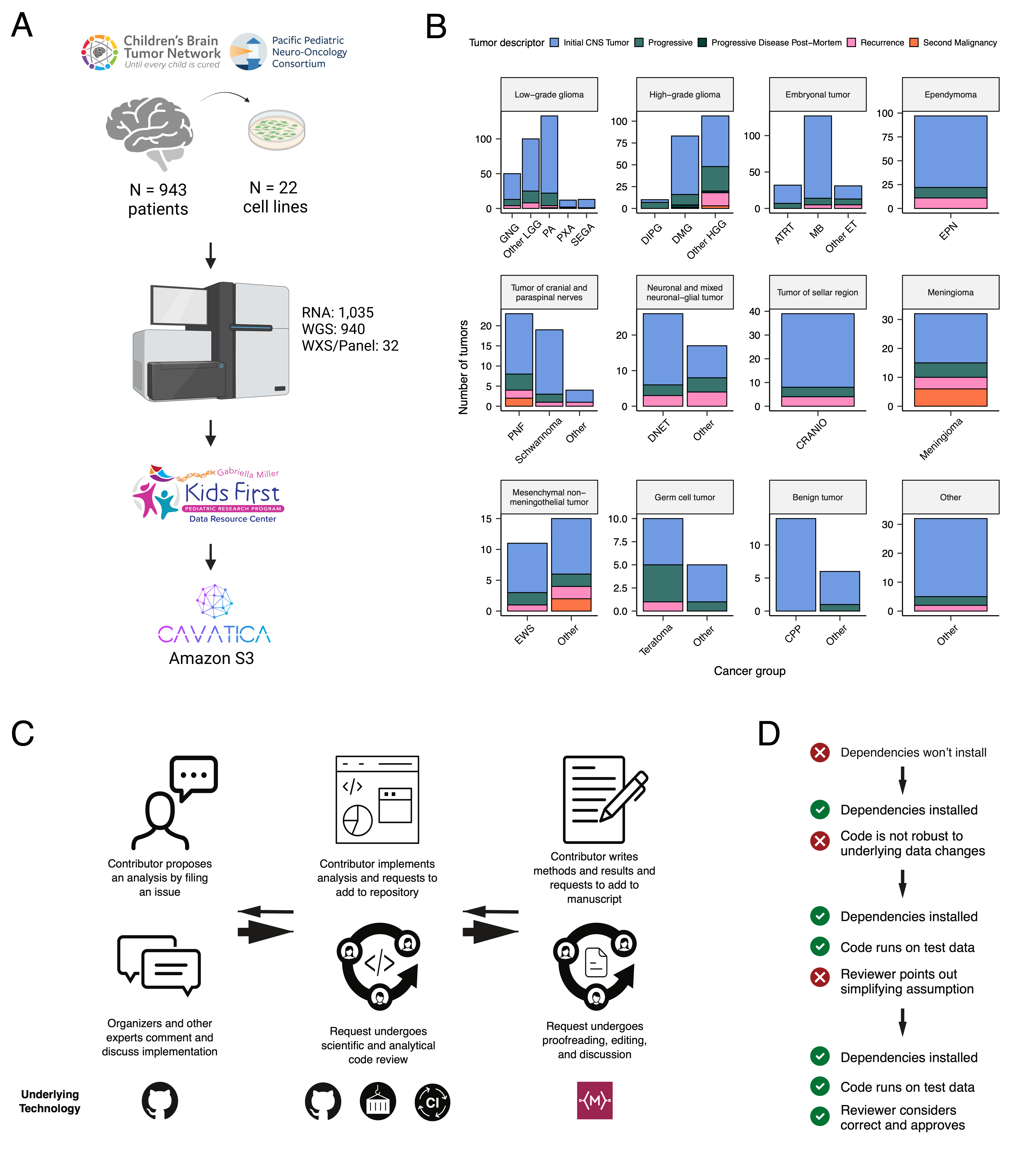
We previously performed whole genome sequencing (WGS), whole exome sequencing (WXS), and RNA sequencing (RNA-Seq) on matched tumor and normal tissues as well as selected cell lines [@doi:10.1093/neuonc/noz192] from 943 patients from the Pediatric Brain Tumor Atlas (PBTA), consisting of 911 patients from the CBTN [@doi:10.1016/j.neo.2022.100846] and 32 patients from PNOC [@doi:10.1002/ijc.32258; @doi:10.1158/1078-0432.CCR-22-0803] (Figure {@fig:Fig1}A). Figure {@fig:Fig1}B summarizes biospecimen numbers by phase of therapy and histology. We harnessed, and built upon, the benchmarking efforts of the Gabriella Miller Kids First Data Resource Center to develop robust and reproducible data analysis workflows within the CAVATICA platform to perform comprehensive somatic analyses (Figure {@fig:S1}) and STAR Methods) of the PBTA.
A key innovative feature of OpenPBTA has been its open contribution framework used for both analyses (i.e., analytical code) and manuscript writing, a model which can be utilized by code contributors within individual scientific groups and/or collaboration. We created a public Github analysis repository (https://github.com/AlexsLemonade/OpenPBTA-analysis) to hold all analysis code downstream of Kids First workflows and a GitHub manuscript repository (https://github.com/AlexsLemonade/OpenPBTA-manuscript) with Manubot [@doi:10.1371/journal.pcbi.1007128] integration to enable real-time manuscript creation. With few exceptions noted in their respective READMEs, analysis modules can be run locally and can easily be scaled and/or run on an Amazon EC2 instance since they are run in the project Docker® [@https://dl.acm.org/doi/10.5555/2600239.2600241] container. Importantly, all analyses and manuscript writing were conducted openly throughout the research project, allowing any researcher in the world an opportunity to contribute.
The process for analysis and manuscript contributions is outlined in Figure {@fig:Fig1}C. First, a potential contributor proposed an analysis by filing an issue in the GitHub analysis repository. Next, project organizers or other contributors with expertise provided feedback about the proposed analysis (Figure {@fig:Fig1}C). The contributor then made a copy (fork) of the analysis repository, to which they added their proposed code and results. The contributor formally requested inclusion of their analytical code and results to the main OpenPBTA analysis repository by filing a pull request (PR) on GitHub. All PRs to the analysis repository underwent peer review by organizers and/or other contributors to ensure scientific accuracy, maintainability, and readability of code and documentation (Figure {@fig:Fig1}C-D). Importantly, this peer review process entailed two or more analysts running the same code within the same Docker® [@https://dl.acm.org/doi/10.5555/2600239.2600241] container to ensure reproducibility of results derived from a specific data release.
The collaborative nature of the project required additional steps beyond peer review of analytical code to ensure consistent results for all collaborators over time (Figure {@fig:Fig1}D). We leveraged Docker® [@https://dl.acm.org/doi/10.5555/2600239.2600241] and the Rocker project [@https://doi.org/10.48550/arXiv.1710.03675] to maintain a consistent software development environment, creating a monolithic image that contained all dependencies necessary for analyses. To ensure that new code would execute in the development environment, we used the continuous integration (CI) service CircleCI® to run analytical code on a subset of data for testing before formal code review, allowing us to detect code bugs or sensitivity to data changes.
We followed a similar process in our Manubot-powered [@doi:10.1371/journal.pcbi.1007128] manuscript repository for additions to the manuscript (Figure {@fig:Fig1}C). Similarly, PRs underwent peer review for clarity and correctness, agreement with interpretation, and spell-checking via Manubot.
 {#fig:Fig1 width="7in"}
{#fig:Fig1 width="7in"}
Over the past two decades, experts in neuro-oncology have worked with the WHO to iteratively redefine the classifications of central nervous system (CNS) tumors [@pubmed:11895036; @doi:10.1007/s00401-007-0243-4].
In 2016 [@doi:10.1007/s00401-016-1545-1], molecular subtypes driven by genetic alterations have been integrated into these classifications.
Since CBTN specimen collection began in 2011 before molecular data were integrated into classifications, the majority of OpenPBTA tumors lacked molecular subtype information at the time of tissue collection.
Moreover, the OpenPBTA does not yet feature methylation arrays which are increasingly used to inform molecular subtyping and cancer diagnosis.
Therefore, we created analysis modules to systematically consider key genomic features of tumor entities described by the WHO in 2016 or Ryall and colleagues [@doi:10.1016/j.ccell.2020.03.011] for low-grade gliomas (LGGs).
Coupled with clinician and pathologist review, we generated research-grade integrated diagnoses for 60% (644/1074) of tumors with high confidence (Table S1) without the requirement for methylation, a major innovation of this project.
This allowed us to align OpenPBTA specimen diagnoses with WHO classifications (e.g., tumors formerly ascribed primitive neuro-ectodermal tumor [PNET] diagnoses), discover rarer tumor entities (e.g., H3-mutant ependymoma, meningioma with YAP1::FAM118B fusion), as well as identify and correct data entry errors (e.g., an ETMR incorrectly entered as a medulloblastoma) and histologically mis-identified specimens (e.g., Ewing sarcoma sample labeled as a craniopharyngioma).
Uniquely, we used transcriptomic classification to subtype 122 medulloblastomas into SHH, WNT, Group 3, or Group 4 with MedulloClassifier [@doi:10.1371/journal.pcbi.1008263] and MM2S [@doi:10.1186/s13029-016-0053-y], achieving accuracies of 95% (41/43) and 91% (39/43), respectively.
Table {@tbl:Table1} lists the number of tumors subtyped within OpenPBTA, comprising LGGs (N = 290), HGGs (N = 141), embryonal tumors (N = 126), ependymomas (N = 33), tumors of sellar region (N = 27), mesenchymal non-meningothelial tumors (N = 11), glialneuronal tumors (N = 10), and chordomas (N = 6), where Ns represent unique tumors. For detailed methods, see STAR Methods and Figure {@fig:S1}.
| Broad histology group | OpenPBTA molecular subtype | Patients | Tumors |
|---|---|---|---|
| Chordoma | CHDM, conventional | 2 | 2 |
| Chordoma | CHDM, poorly differentiated | 2 | 4 |
| Embryonal tumor | CNS Embryonal, NOS | 13 | 13 |
| Embryonal tumor | CNS HGNET-MN1 | 1 | 1 |
| Embryonal tumor | CNS NB-FOXR2 | 2 | 3 |
| Embryonal tumor | ETMR, C19MC-altered | 5 | 5 |
| Embryonal tumor | ETMR, NOS | 1 | 1 |
| Embryonal tumor | MB, Group3 | 14 | 14 |
| Embryonal tumor | MB, Group4 | 48 | 49 |
| Embryonal tumor | MB, SHH | 24 | 30 |
| Embryonal tumor | MB, WNT | 10 | 10 |
| Ependymoma | EPN, H3 K28 | 1 | 1 |
| Ependymoma | EPN, ST RELA | 25 | 28 |
| Ependymoma | EPN, ST YAP1 | 3 | 4 |
| High-grade glioma | DMG, H3 K28 | 18 | 24 |
| High-grade glioma | DMG, H3 K28, TP53 activated | 10 | 13 |
| High-grade glioma | DMG, H3 K28, TP53 loss | 30 | 40 |
| High-grade glioma | HGG, H3 G35 | 3 | 3 |
| High-grade glioma | HGG, H3 G35, TP53 loss | 1 | 1 |
| High-grade glioma | HGG, H3 wildtype | 26 | 31 |
| High-grade glioma | HGG, H3 wildtype, TP53 activated | 5 | 5 |
| High-grade glioma | HGG, H3 wildtype, TP53 loss | 14 | 21 |
| High-grade glioma | HGG, IDH, TP53 activated | 1 | 2 |
| High-grade glioma | HGG, IDH, TP53 loss | 1 | 1 |
| Low-grade glioma | GNG, BRAF V600E | 13 | 13 |
| Low-grade glioma | GNG, BRAF V600E, CDKN2A/B | 1 | 1 |
| Low-grade glioma | GNG, FGFR | 1 | 1 |
| Low-grade glioma | GNG, H3 | 1 | 1 |
| Low-grade glioma | GNG, IDH | 1 | 2 |
| Low-grade glioma | GNG, KIAA1549-BRAF | 5 | 5 |
| Low-grade glioma | GNG, MYB/MYBL1 | 1 | 1 |
| Low-grade glioma | GNG, NF1-germline | 1 | 1 |
| Low-grade glioma | GNG, NF1-somatic, BRAF V600E | 1 | 1 |
| Low-grade glioma | GNG, other MAPK | 4 | 4 |
| Low-grade glioma | GNG, other MAPK, IDH | 1 | 1 |
| Low-grade glioma | GNG, RTK | 2 | 3 |
| Low-grade glioma | GNG, wildtype | 14 | 14 |
| Low-grade glioma | LGG, BRAF V600E | 25 | 27 |
| Low-grade glioma | LGG, BRAF V600E, CDKN2A/B | 5 | 5 |
| Low-grade glioma | LGG, FGFR | 8 | 8 |
| Low-grade glioma | LGG, IDH | 3 | 3 |
| Low-grade glioma | LGG, KIAA1549-BRAF | 106 | 113 |
| Low-grade glioma | LGG, KIAA1549-BRAF, NF1-germline | 1 | 1 |
| Low-grade glioma | LGG, KIAA1549-BRAF, other MAPK | 1 | 1 |
| Low-grade glioma | LGG, MYB/MYBL1 | 2 | 2 |
| Low-grade glioma | LGG, NF1-germline | 6 | 6 |
| Low-grade glioma | LGG, NF1-germline, CDKN2A/B | 1 | 1 |
| Low-grade glioma | LGG, NF1-germline, FGFR | 1 | 2 |
| Low-grade glioma | LGG, NF1-somatic | 2 | 2 |
| Low-grade glioma | LGG, NF1-somatic, FGFR | 1 | 1 |
| Low-grade glioma | LGG, NF1-somatic, NF1-germline, CDKN2A/B | 1 | 1 |
| Low-grade glioma | LGG, other MAPK | 11 | 12 |
| Low-grade glioma | LGG, RTK | 8 | 10 |
| Low-grade glioma | LGG, RTK, CDKN2A/B | 1 | 1 |
| Low-grade glioma | LGG, wildtype | 33 | 34 |
| Low-grade glioma | SEGA, RTK | 1 | 1 |
| Low-grade glioma | SEGA, wildtype | 10 | 11 |
| Mesenchymal non-meningothelial tumor | EWS | 9 | 11 |
| Neuronal and mixed neuronal-glial tumor | CNC | 2 | 2 |
| Neuronal and mixed neuronal-glial tumor | EVN | 1 | 1 |
| Neuronal and mixed neuronal-glial tumor | GNT, BRAF V600E | 1 | 1 |
| Neuronal and mixed neuronal-glial tumor | GNT, KIAA1549-BRAF | 1 | 2 |
| Neuronal and mixed neuronal-glial tumor | GNT, other MAPK | 1 | 1 |
| Neuronal and mixed neuronal-glial tumor | GNT, other MAPK, FGFR | 1 | 1 |
| Neuronal and mixed neuronal-glial tumor | GNT, RTK | 1 | 2 |
| Tumor of sellar region | CRANIO, ADAM | 27 | 27 |
| Total | 577 | 644 |
Table: Molecular subtypes generated through the OpenPBTA project. Listed are broad tumor histologies, molecular subtypes generated, and number of patients and tumors subtyped within OpenPBTA. {#tbl:Table1}
We performed a comprehensive genomic analysis of somatic SNVs, CNVs, SVs, and fusions across 1,074 tumors (N = 1,019 RNA-Seq, N = 918 WGS, N = 32 WXS/Panel) and 22 cell lines (N = 16 RNA-Seq, N = 22 WGS), from 943 patients, 833 with paired normal specimens (N = 801 WGS, N = 32 WXS/Panel). Tumor purity across PBTA samples was high (median of 76%), though we observe cancer groups with lower purity: SEGA, PXA, and teratoma (Figure {@fig:S3}A). Unless otherwise noted, each analysis was performed for diagnostic tumors using one tumor per patient.
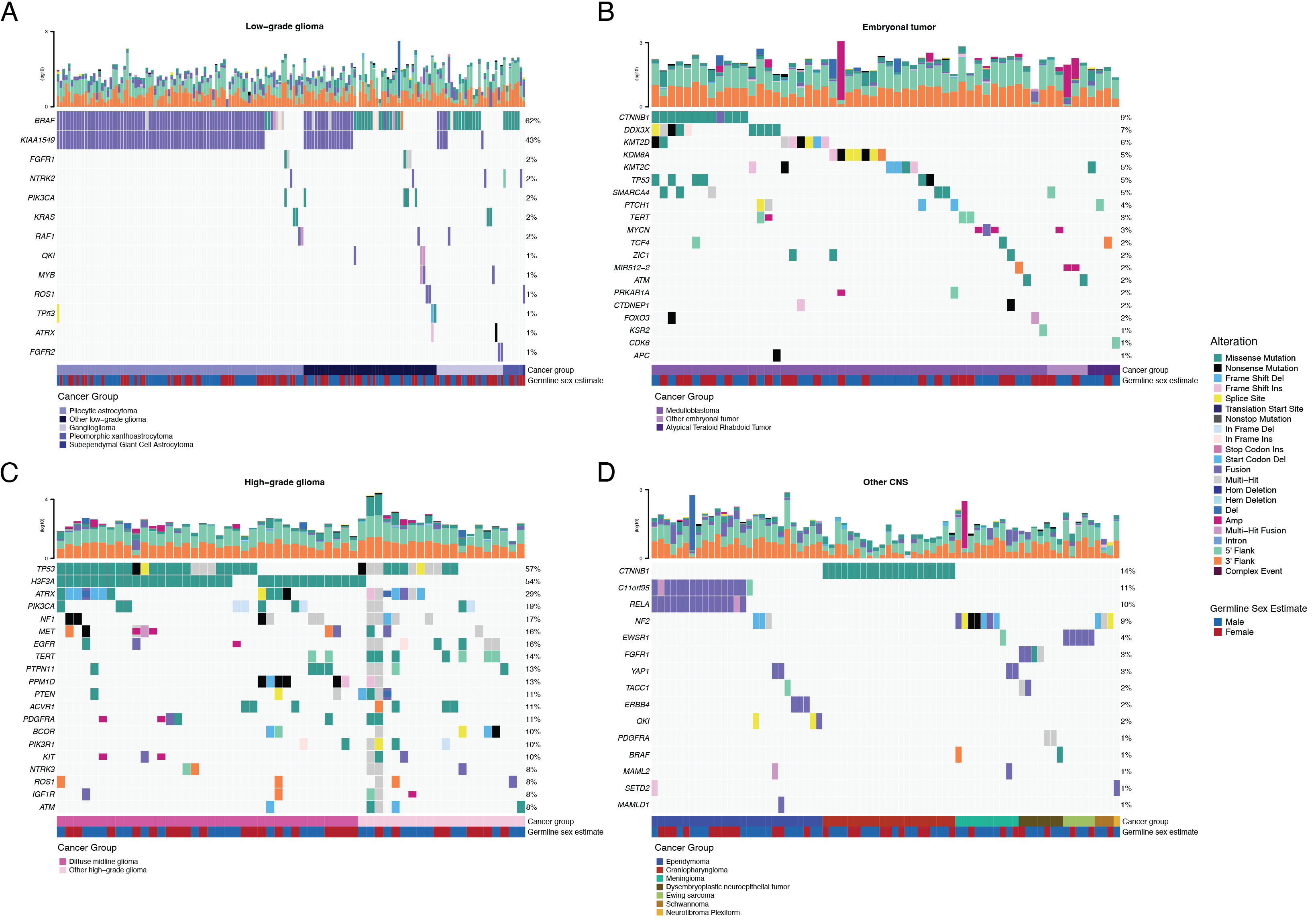
Following SNV consensus calling (Figure {@fig:S1} and Figure {@fig:S2}A-G), we observed as expected lower tumor mutation burden (TMB) Figure {@fig:S2}H in pediatric tumors compared to adult brain tumors from The Cancer Genome Atlas (TCGA), Figure {@fig:S2}I, with hypermutant (> 10 Mut/Mb) and ultra-hypermutant (> 100 Mut/Mb) tumors [@doi:10.1016/j.cell.2017.09.048] only found within HGGs. Figure {@fig:Fig2} and Figure {@fig:S3}A depict oncoprints recapitulating known histology-specific driver genes in primary tumors across OpenPBTA histologies, and Table S2 summarizes all detected alterations across cancer groups.
As expected, the majority (62%, 140/226) of LGGs harbored a somatic alteration in BRAF, with canonical BRAF::KIAA1549 fusions as the major oncogenic driver [@doi:10.1186/s40478-020-00902-z] (Figure {@fig:Fig2}A). We observed additional mutations in FGFR1 (2%), PIK3CA (2%), KRAS (2%), TP53 (1%), and ATRX (1%) and fusions in NTRK2 (2%), RAF1 (2%), MYB (1%), QKI (1%), ROS1 (1%), and FGFR2 (1%), concordant with previous studies reporting near universal upregulation of the RAS/MAPK pathway in these tumors resulting from activating mutations and/or oncogenic fusions [@doi:10.1186/s40478-020-00902-z; @doi:10.1016/j.ccell.2020.03.011]. Indeed, we observed significant upregulation (ANOVA Bonferroni-corrected p < 0.01) of the KRAS signaling pathway in LGGs (Figure {@fig:Fig5}B) using gene set variant analysis (GSVA).
The majority (N = 95) of embryonal tumors were medulloblastomas that spanned the spectrum of molecular subtypes (WNT, SHH, Group3, and Group 4; see Molecular Subtyping of CNS Tumors), as identified by subtype-specific canonical mutations (Figure {@fig:Fig2}B). We detected canonical SMARCB1/SMARCA4 deletions or inactivating mutations in atypical teratoid rhabdoid tumors (ATRTs; shown in Table S2) and C19MC amplification in the embryonal tumors with multilayer rosettes (ETMRs, displayed within "Other embryonal tumors" in Figure {@fig:Fig2}B) [@doi:10.1007/s00401-020-02182-2; @doi:10.1093/neuonc/noab178; @doi:10.1186/s40478-020-00984-9; @doi:10.1038/nature22973].
Across HGGs, we found that TP53 (57%, 36/63) and H3F3A (54%, 34/63) were both most mutated and co-occurring genes (Figure {@fig:Fig2}A and C), followed by frequent mutations in ATRX (29%, 18/63), a gene commonly mutated in gliomas [@doi:10.1080/14728222.2018.1487953]. We observed recurrent amplifications and fusions in EGFR, MET, PDGFRA, and KIT, highlighting the multiple oncogenic mechanisms these tumors utilize to activate tyrosine kinases, as previously reported [@doi:10.1002/ijc.32258; @doi:10.1016/j.ccell.2017.08.017; @doi:10.1186/s40478-020-00905-w]. GSVA showed upregulation (ANOVA Bonferroni-corrected p < 0.01) of DNA repair, G2M checkpoint, and MYC pathways as well as downregulation of the TP53 pathway (Figure {@fig:Fig5}B). The two tumors with ultra-high TMB (> 100 Mutations/Mb) were from patients with known mismatch repair deficiency syndrome [@doi:10.1093/neuonc/noz192].
We observed that 25% (15/60) of ependymomas were C11orf95::RELA (now, ZFTA::RELA) fusion-positive ependymomas [@doi:10.1038/nature13109] and that 68% (21/31) of craniopharyngiomas were driven by mutations in CTNNB1 (Figure {@fig:Fig2}D). Multiple histologies contained somatic mutations or fusions in NF2, including 41% (7/17) of meningiomas, 5% (3/60) of ependymomas, and 25% (3/12) of schwannomas. We observed rare fusions in ERBB4, YAP1, and/or QKI in 10% (6/60) of ependymomas. DNETs harbored alterations in MAPK/PI3K pathway genes, as has been previously reported [@doi:10.1093/jnen/nlz101], including FGFR1 (21%, 4/19), PDGFRA (10%, 2/19), and BRAF (5%, 1/19). Frequent mutations in additional rare brain tumor histologies are depicted in Figure {@fig:S3}A.
 {#fig:Fig2 width="9in"}
{#fig:Fig2 width="9in"}
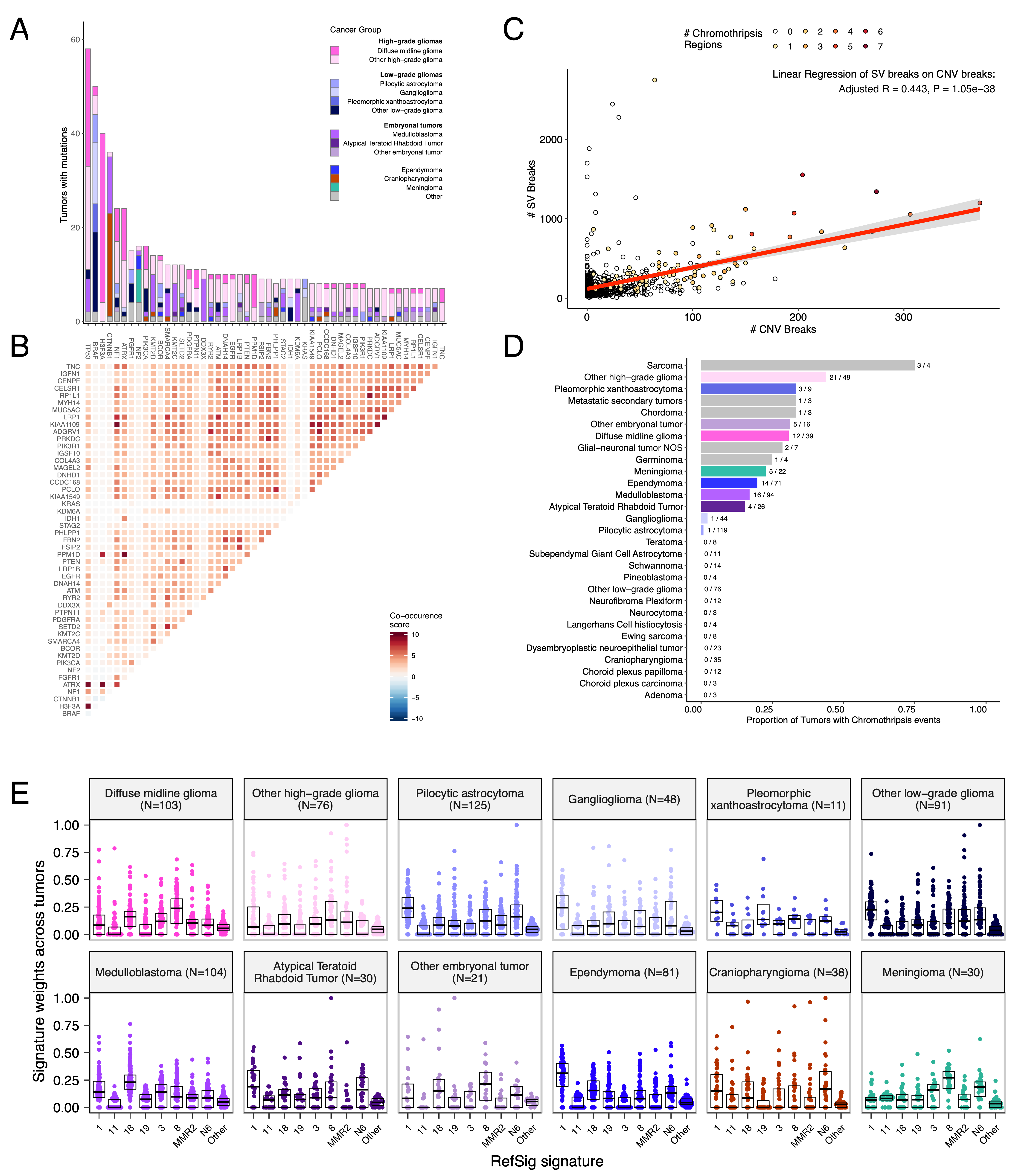
We analyzed mutational co-occurrence across the OpenPBTA, using a single tumor from each patient with available WGS (N = 668 patients). The top 50 mutated genes (see STAR Methods for details) in primary tumors are shown in Figure {@fig:Fig3} by tumor type (A, bar plots), with co-occurrence scores illustrated in the heatmap (B). As expected, TP53 was the most frequently mutated gene across the OpenPBTA (8.7%, 58/668), significantly co-occurring with H3F3A (OR = 30.05, 95% CI: 14.5 - 62.3, q = 2.34e-16), ATRX (OR = 23.3, 95% CI: 9.6 - 56.3, q = 8.72e-9), NF1 (OR = 8.26, 95% CI: 3.5 - 19.4, q = 7.40e-5), and EGFR (OR = 17.5, 95% CI: 4.8 - 63.9, q = 2e-4), with all of these driven by HGGs and consistent with previous reports [@doi:10.1016/j.ccell.2017.08.017; @doi:10.1093/neuonc/noaa251; @doi:10.1038/ng.2938].
In embryonal tumors, mutations in CTNNB1 significantly co-occurred with mutations in TP53 (OR = 43.6 95% CI: 7.1 - 265.8, q = 1.52e-3) as well as with mutations in DDX3X (OR = 21.4, 95% CI: 4.7 - 97.9, q = 4.15e-3), events driven by medulloblastomas as previously reported [@doi:10.1038/nrc3410; @doi:10.1200/JCO.2010.31.1670]. Mutations in FGFR1 and PIK3CA significantly co-occurred in LGGs (OR = 77.25, 95% CI: 10.0 - 596.8, q = 3.12e-3), consistent with previous findings [@doi:10.1200/JCO.2010.31.1670; @doi:10.1186/s40478-020-01027-z]. Of HGG tumors with mutations in TP53 or PPM1D, 53/55 (96.3%) had mutations in only one of these genes (OR = 0.17, 95% CI: 0.04 - 0.89, q = 0.056). This trend recapitulates previous observations that TP53 and PPM1D mutations tend to be mutually exclusive in HGGs [@https://doi.org/10.1038/ng.2938].
We summarized broad CNVs and SVs and observed that HGGs and DMGs, followed by medulloblastomas, had the most unstable genomes (Figure {@fig:S3}C). By contrast, craniopharyngiomas and schwannomas generally lacked somatic CNV. Together, these CNV patterns largely aligned with our estimates of tumor mutational burden (Figure {@fig:S2}H). Breakpoint density estimated from SV and CNV data was significantly correlated across tumors (linear regression p = 1.05e-58) (Figure {@fig:Fig3}C) and as expected, the number of chromothripsis regions called increased as breakpoint density increased (Figure {@fig:S3}D-E). We identified chromothripsis events in 31% (N = 12/39) of DMGs and in 44% (N = 21/48) of other HGGs (Figure {@fig:Fig3}D). We also found evidence of chromothripsis in over 15% of sarcomas, PXAs, metastatic secondary tumors, chordomas, glial-neuronal tumors, germinomas, meningiomas, ependymomas, medulloblastomas, ATRTs, and other embryonal tumors, highlighting the genomic instability and complexity of these pediatric brain tumors.
We next assessed the contributions of eight previously identified adult CNS-specific mutational signatures from the RefSig database [@doi:10.1038/s43018-020-0027-5] across tumors (Figure {@fig:Fig3}E and Figure {@fig:S4}A). Stage 0 and/or 1 tumors characterized by low TMBs (Figure {@fig:S2}H) such as pilocytic astrocytomas, gangliogliomas, other LGGs, and craniopharyngiomas, were dominated by Signature 1 (Figure {@fig:S4}A), which results from the normal spontaneous deamination of 5-methylcytosine process. Signature N6 is a CNS-specific signature which we observed almost universally across tumors. Drivers of Signature 18, TP53, APC, NOTCH1 (found at https://signal.mutationalsignatures.com/explore/referenceCancerSignature/31/drivers), are also canonical drivers of medulloblastoma, and indeed, we observed Signature 18 as the signature with the highest weight in medulloblastoma tumors. Signatures 3, 8, 18, and MMR2 were prevalent in HGGs, including DMGs. Finally, we found that the Signature 1 weight was higher at diagnosis (pre-treatment) and was almost always lower in tumors at later phases of therapy (progression, recurrence, post-mortem, secondary malignancy (Figure {@fig:S4}B). This trend may have resulted from therapy-induced mutations that produced additional signatures (e.g., temozolomide treatment has been suggested to drive Signature 11 [@doi:10.1053/j.gastro.2014.07.052]), subclonal expansion, and/or acquisition of additional driver mutations during tumor progression, leading to higher overall TMBs and additional signatures.
 {#fig:Fig3 width="7in"}
{#fig:Fig3 width="7in"}
The majority of RNA-Seq samples in the PBTA cohort were prepared with ribosomal RNA depletion followed by stranded sequencing (N = 977), while the remaining samples were prepared with poly-A selection (N = 58). Since batch correction was not feasible (see Limitations of the Study and Figure {@fig:S7}A), the following analyses were performed using stranded samples only.
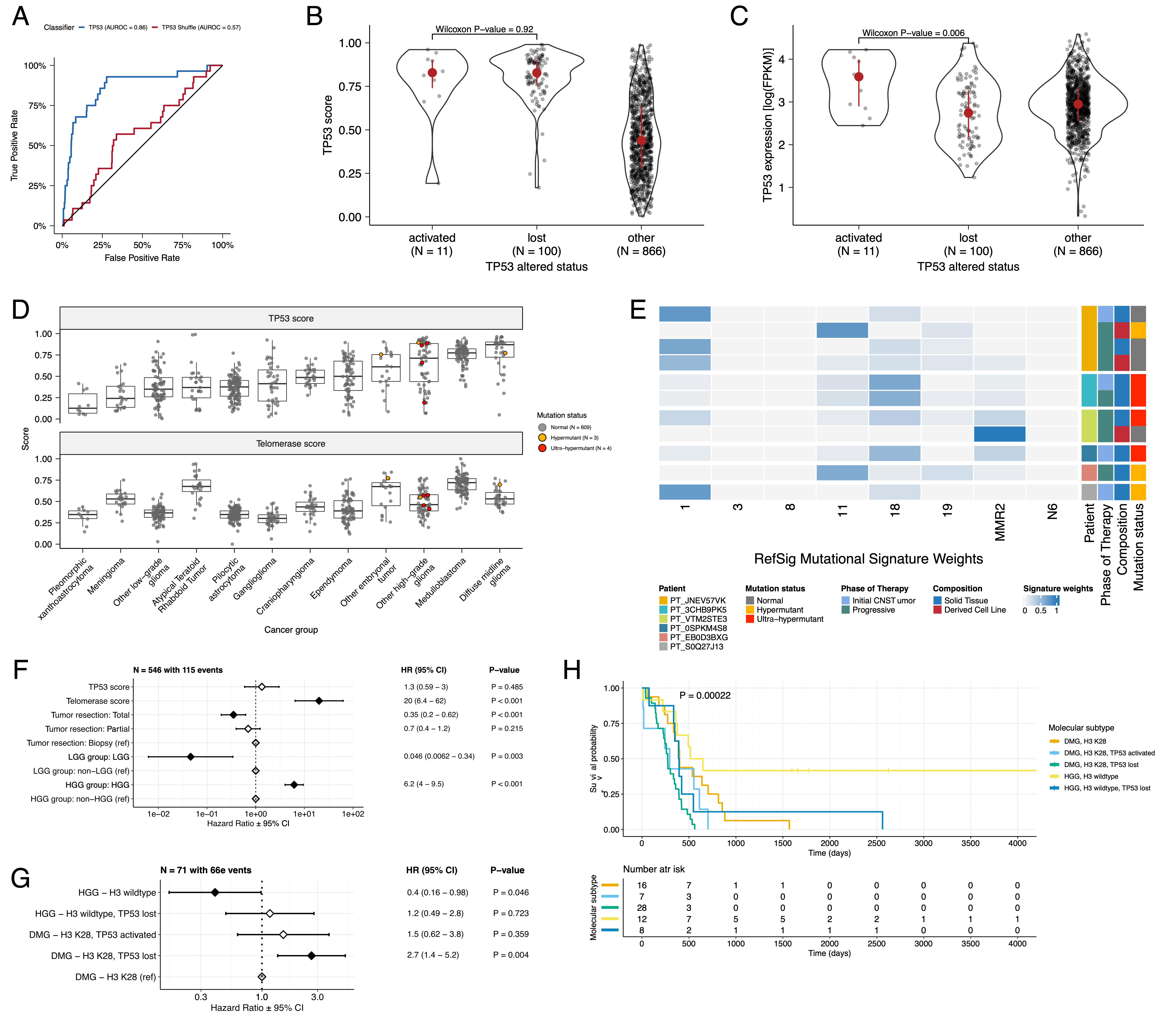
To understand the TP53 phenotype in each tumor, we ran a classifier previously trained on TCGA [@doi:10.1016/j.celrep.2018.03.076] to calculate a TP53 score and infer TP53 inactivation status. We identified "true positive" TP53 alterations derived using high-confidence SNVs, CNVs, SVs, and fusions in TP53 and annotated tumors as "activated" if they harbored one of p.R273C or p.R248W gain-of-function mutations [@doi:10.1038/ng0593-42], or "lost" if the given patient had 1) a Li Fraumeni Syndrome (LFS) predisposition diagnosis, 2) the tumor harbored a known hotspot mutation, or 3) the tumor contained two hits (e.g. both SNV and CNV), suggesting both alleles were affected. If the TP53 mutation did not reside within the DNA-binding domain or we did not detect any alteration in TP53, we annotated the tumor as "other," reflecting an unknown TP53 alteration status. The classifier achieved a high accuracy (AUROC = 0.86) for rRNA-depleted, stranded tumors compared to randomly shuffled TP53 scores (Figure {@fig:Fig4}A). By contrast, while this classifier has previously shown strong performance on poly-A data from both adult [@doi:10.1016/j.celrep.2018.03.076] tumors and pediatric patient-derived xenografts [@doi:10.1016/j.celrep.2019.09.071], it did not perform as well on the poly-A tumors in this cohort (AUROC = 0.62; Figure {@fig:S5}A).
While we expected that tumors annotated as "lost" would have higher TP53 scores than would tumors annotated as "other," we observed that tumors annotated as "activated" had similar TP53 scores to those annotated as "lost" (Figure {@fig:Fig4}B, Wilcoxon p = 0.92). This result suggests that the classifier actually detects an oncogenic, or altered, TP53 phenotype (scores > 0.5) rather than solely TP53 inactivation, as interpreted previously [@doi:10.1016/j.celrep.2018.03.076]. Moreover, tumors with "activating" TP53 mutations showed higher TP53 expression compared to those with TP53 "loss" mutations (Wilcoxon p = 0.006, Figure {@fig:Fig4}C). Tumor types with the highest median TP53 scores were those known to harbor somatic TP53 alterations and included DMGs, medulloblastomas, HGGs, DNETs, ependymomas, and craniopharyngiomas (Figure {@fig:Fig4}D), while gangliogliomas, LGGs, meningiomas, and schwannomas had the lowest median scores.
To further validate the classifier's accuracy, we assessed TP53 scores for patients with LFS, hypothesizing that all of these tumors would have high scores. Indeed, we observed higher scores in 8/10 tumors from LFS patients (N = 8 patients)(Table S3). Although we observed low TP53 scores in two tumors from LFS patients (BS_DEHJF4C7 with a score of 0.09 and BS_ZD5HN296 with a score of 0.28), we confirmed from pathology reports that both patients were diagnosed with LFS and had a pathogenic germline variant in TP53. In addition, the tumor purity of these two LFS tumors was low (16% and 37%, respectively), suggesting the classifier may require a certain level of tumor purity to achieve good performance, as we expect TP53 to be intact in normal cells. We posit that these transcriptomic scores can be utilized to infer TP53 function in the absence of a predicted oncogenic TP53 alteration or DNA sequencing in general.
We used gene expression data to predict telomerase activity using EXpression-based Telomerase ENzymatic activity Detection (EXTEND) [@doi:10.1038/s41467-020-20474-9] as a surrogate measure of malignant potential [@doi:10.1038/s41467-020-20474-9; @doi:10.1093/carcin/bgp268], such that higher EXTEND scores infer higher telomerase activity.
While we did not find that tumors with TERT promoter (TERTp) mutations (N = 6) had significantly higher telomerase activity scores than non-mutated tumors (Wilcoxon p-value = 0.1196), we observed that EXTEND scores significantly correlated with TERC (R = 0.619, p < 0.01) and TERT (R = 0.491, p < 0.01) expression (Figure {@fig:S5}B-C).
Since catalytically-active telomerase requires a combination of full-length TERT, TERC, as well as accessory proteins [@url:https://pubmed.ncbi.nlm.nih.gov/9751630], we expect that EXTEND scores may not be exclusively correlated with TERT alterations and expression.
Next, we found aggressive tumors such as HGGs (DMGs and other HGGs) and MB had high EXTEND scores (Figure {@fig:Fig4}D), while low-grade lesions such as schwannomas, GNGs, DNETs, and other LGGs had among the lowest scores (Table S3).
These findings support previous reports of a more aggressive phenotype in tumors with higher telomerase activity [@doi:10.1007/s13277-016-5045-7; @doi:10.1038/labinvest.3700710; @doi:10.1007/s12032-016-0736-x; @doi:10.1111/j.1750-3639.2010.00372.x].
We further investigated the mutational signature profiles of hypermutant (TMB > 10 Mut/Mb; N = 3) and ultra-hypermutant (TMB > 100 Mut/Mb; N = 4) tumors and/or derived cell lines from six patients in OpenPBTA (Figure {@fig:Fig4}E). Five of six tumors were diagnosed as HGGs and one was a brain metastasis of a MYCN non-amplified neuroblastoma tumor. Signature 11, which is associated with exposure to temozolomide plus MGMT promoter and/or mismatch repair deficiency [@doi:10.1038/s41588-019-0525-5], was indeed present in tumors with previous exposure to the drug (Table {@tbl:Table2}). We detected the MMR2 signature in tumors of four patients (PT_0SPKM4S8, PT_3CHB9PK5, PT_JNEV57VK, and PT_VTM2STE3) diagnosed with either constitutional mismatch repair deficiency (CMMRD) or Lynch syndrome (Table {@tbl:Table2}), genetic predisposition syndromes caused by a variant in a mismatch repair gene such as PMS2, MLH1, MSH2, MSH6, or others [@doi:10.1136/jmedgenet-2020-107627]. Three of these patients harbored pathogenic germline variants in one of the aforementioned genes. While we did not find a known pathogenic variant in the germline of PT_VTM2STE3, this patient had a self-reported PMS2 variant noted in their pathology report and we did find 19 intronic variants of unknown significance (VUS) in PMS2. This is not surprising since an estimated 49% of germline PMS2 variants in patients with CMMRD and/or Lynch syndrome are VUS [@doi:10.1136/jmedgenet-2020-107627]. Interestingly, while the cell line derived from patient PT_VTM2STE3's tumor at progression was not hypermutated (TMB = 5.7 Mut/Mb), it solely showed the MMR2 signature of the eight CNS signatures examined, suggesting selective pressure to maintain a mismatch repair (MMR) phenotype in vitro. From patient PT_JNEV57VK, only one of the two cell lines derived from the progressive tumor was hypermutated (TMB = 35.9 Mut/Mb). This hypermutated cell line was strongly weighted towards signature 11, while this patient's non-hypermutated cell line showed several lesser signature weights (1, 11, 18, 19, MMR2; Table S2), highlighting the plasticity of mutational processes and the need to carefully genomically characterize and select models for preclinical studies based on research objectives.
Signature 18, which has been associated with high genomic instability and can lead to a hypermutator phenotype [@doi:10.1038/s43018-020-0027-5], was uniformly represented among hypermutant solid tumors. Additionally, we found that all of the HGG tumors or cell lines had dysfunctional TP53 (Table {@tbl:Table2}), consistent with a previous report showing TP53 dysregulation is a dependency in tumors with high genomic instability [@doi:10.1038/s43018-020-0027-5]. With one exception, hypermutant and ultra-hypermutant tumors had high TP53 scores (> 0.5) and telomerase activity. Interestingly, none of the hypermutant tumors showed evidence of signature 3 (present in homologous recombination deficient tumors), signature 8 (arises from double nucleotide substitutions/unknown etiology), or signature N6 (a universal CNS tumor signature). The mutual exclusivity of signatures 3 and MMR2 corroborates a previous report suggesting tumors do not tend to feature both deficient homologous repair and mismatch repair [@doi:10.1016/j.celrep.2018.03.076].
| Kids First Participant ID | Kids First Biospecimen ID | CBTN ID | Phase of therapy | Composition | Therapy post-biopsy | Cancer predisposition | Pathogenic germline variant | TMB | OpenPBTA molecular subtype |
|---|---|---|---|---|---|---|---|---|---|
| PT_0SPKM4S8 | BS_VW4XN9Y7 | 7316-2640 | Initial CNS Tumor | Solid Tissue | Radiation, Temozolomide, CCNU | None documented | NM_000535.7(PMS2):c.137G>T (p.Ser46Ile) (LP) | 187.4 | HGG, H3 wildtype, TP53 activated |
| PT_3CHB9PK5 | BS_20TBZG09 | 7316-515 | Initial CNS Tumor | Solid Tissue | Radiation, Temozolomide, Irinotecan, Bevacizumab | CMMRD | NM_000179.3(MSH6):c.3439-2A>G (LP) | 307 | HGG, H3 wildtype, TP53 loss |
| PT_3CHB9PK5 | BS_8AY2GM4G | 7316-2085 | Progressive | Solid Tissue | Radiation, Temozolomide, Irinotecan, Bevacizumab | CMMRD | NM_000179.3(MSH6):c.3439-2A>G (LP) | 321.6 | HGG, H3 wildtype, TP53 loss |
| PT_EB0D3BXG | BS_F0GNWEJJ | 7316-3311 | Progressive | Solid Tissue | Radiation, Nivolumab | None documented | None detected | 26.3 | Metastatic NBL, MYCN non-amplified |
| PT_JNEV57VK | BS_85Q5P8GF | 7316-2594 | Initial CNS Tumor | Solid Tissue | Radiation, Temozolomide | Lynch Syndrome | NM_000251.3(MSH2):c.1906G>C (p.Ala636Pro) (P) | 4.7 | DMG, H3 K28, TP53 loss |
| PT_JNEV57VK | BS_HM5GFJN8 | 7316-3058 | Progressive | Derived Cell Line | Radiation, Temozolomide, Nivolumab | Lynch Syndrome | NM_000251.3(MSH2):c.1906G>C (p.Ala636Pro) (P) | 35.9 | DMG, H3 K28, TP53 loss |
| PT_JNEV57VK | BS_QWM9BPDY | 7316-3058 | Progressive | Derived Cell Line | Radiation, Temozolomide, Nivolumab | Lynch Syndrome | NM_000251.3(MSH2):c.1906G>C (p.Ala636Pro) (P) | 7.4 | DMG, H3 K28, TP53 loss |
| PT_JNEV57VK | BS_P0QJ1QAH | 7316-3058 | Progressive | Solid Tissue | Radiation, Temozolomide, Nivolumab | Lynch Syndrome | NM_000251.3(MSH2):c.1906G>C (p.Ala636Pro) (P) | 6.3 | DMG, H3 K28, TP53 activated |
| PT_S0Q27J13 | BS_P3PF53V8 | 7316-2307 | Initial CNS Tumor | Solid Tissue | Radiation, Temozolomide, Irinotecan | None documented | None detected | 15.5 | HGG, H3 wildtype, TP53 activated |
| PT_VTM2STE3 | BS_ERFMPQN3 | 7316-2189 | Progressive | Derived Cell Line | Unknown | Lynch Syndrome | None detected | 5.7 | HGG, H3 wildtype, TP53 loss |
| PT_VTM2STE3 | BS_02YBZSBY | 7316-2189 | Progressive | Solid Tissue | Unknown | Lynch Syndrome | None detected | 274.5 | HGG, H3 wildtype, TP53 activated |
Table: Patients with hypermutant tumors. Listed are patients with at least one hypermutant or ultra-hypermutant tumor or cell line. Pathogenic (P) or likely pathogenic (LP) germline variants, coding region TMB, phase of therapy, therapeutic interventions, cancer predisposition (CMMRD = Constitutional mismatch repair deficiency), and molecular subtypes are included. {#tbl:Table2}
Next, we asked whether transcriptomic classification of TP53 dysregulation and/or telomerase activity recapitulate the known prognostic influence of these oncogenic biomarkers.
We identified several expected trends, including a significant overall survival benefit if the tumor had been fully resected (HR = 0.35, 95% CI = 0.2 - 0.62, p < 0.001) or if the tumor belonged to the LGG group (HR = 0.046, 95% CI = 0.0062 - 0.34, p = 0.003) as well as a significant risk if the tumor belonged to the HGG group (HR = 6.2, 95% CI = 4.0 - 9.5, p < 0.001) (Figure {@fig:Fig4}F; STAR Methods).
High telomerase scores were associated with poor prognosis across brain tumor histologies (HR = 20, 95% CI = 6.4 - 62, p < 0.001), demonstrating that EXTEND scores calculated from RNA-Seq are an effective rapid surrogate measure for telomerase activity.
Although higher TP53 scores, which predict TP53 gene or pathway dysregulation, were not a significant predictor of risk across the entire OpenPBTA cohort (Table S4), we did find a significant survival risk associated with higher TP53 scores within DMGs (HR = 6436, 95% CI = 2.67 - 1.55e7, p = 0.03) and ependymomas (HR = 2003, 95% CI = 9.9 - 4.05e5, p = 0.005).
Since we observed the negative prognostic effect of TP53 scores for HGGs, we assessed the effect of molecular subtypes within HGGs on survival risk.
We found that DMG H3 K28 tumors with TP53 loss had significantly worse prognosis (HR = 2.8, CI = 1.4-5.6, p = 0.003) than did DMG H3 K28 tumors with wildtype TP53 (Figure {@fig:Fig4}G and Figure {@fig:Fig4}H).
This finding was also recently reported in two recent restrospective analyses of DIPG tumors [@doi:10.1158/1078-0432.CCR-22-0803; @doi:10.1007/s11060-021-03890-9].
 {#fig:Fig4 width="7in"}
{#fig:Fig4 width="7in"}
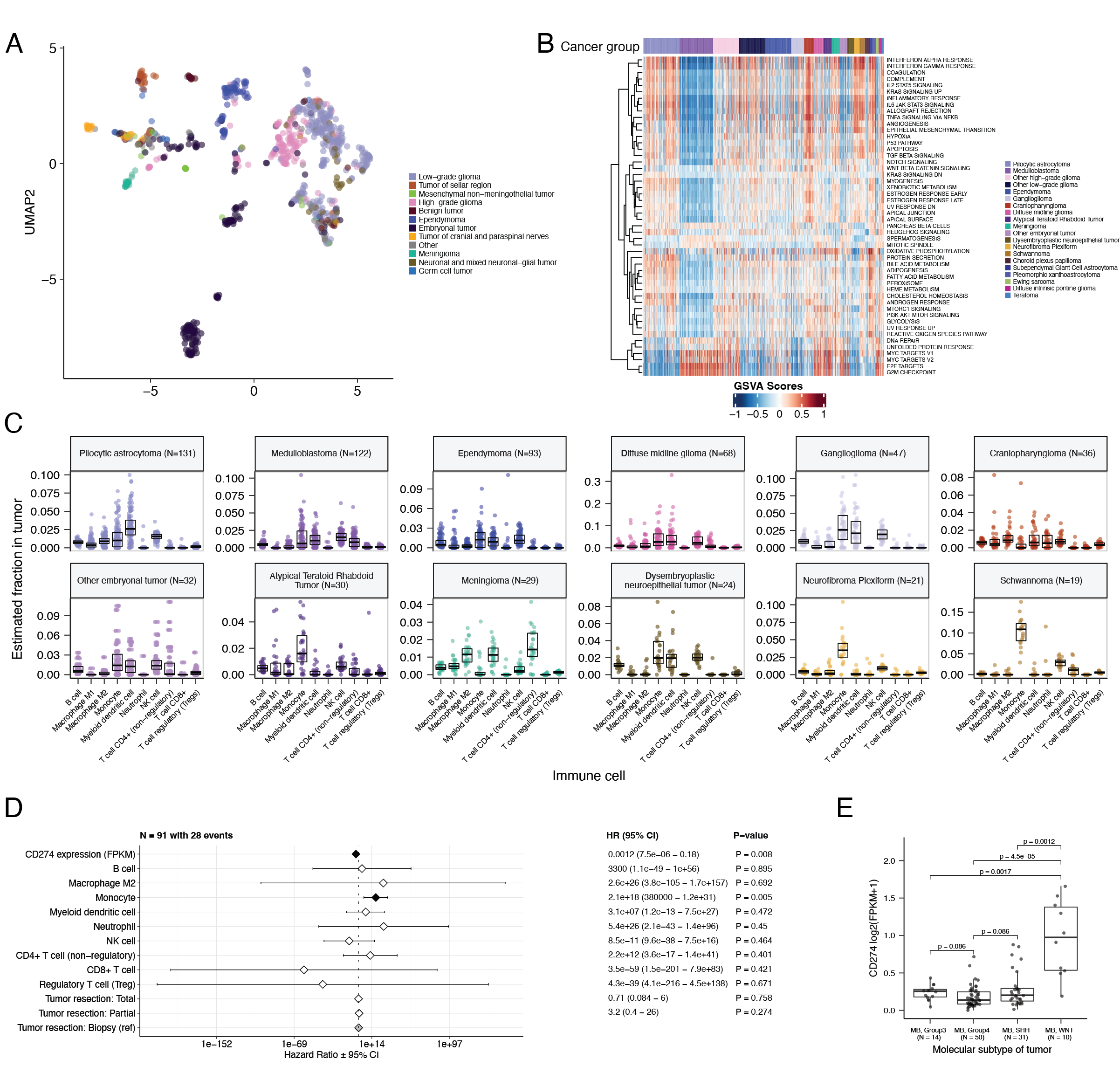
UMAP visualization of gene expression variation across brain tumors (Figure {@fig:Fig5}A) showed expected histological clustering of brain tumors. We further observed that, except for three outliers, C11orf95::RELA (ZFTA::RELA) fusion-positive ependymomas fell within distinct clusters (Figure {@fig:S6}A). Medulloblastoma (MB) tumors cluster by molecular subtype, with WNT and SHH in distinct clusters and Groups 3 and 4 showing some overlap (Figure {@fig:S6}B), as expected. Of note, two MB tumors annotated as the SHH subtype did not cluster with the other MB tumors, and one clustered with Group 3 and 4 tumors, suggesting potential subtype misclassification or different underlying biology of these two tumors. BRAF-driven LGGs (Figure {@fig:S6}C) were present in three separate clusters, suggesting that there might be additional shared biology within each cluster. Histone H3 G35-mutant HGGs generally clustered together and away from K28-mutant tumors (Figure {@fig:S6}D). Interestingly, although H3 K28-mutant tumors have different biological drivers than do H3 wildtype tumors [@doi:10.1126/science.1232245], they did not form distinct clusters. This pattern suggests these subtypes may be driven by common transcriptional programs, have other much stronger biological drivers than their known distinct epigenetic drivers, or our sample size is too small to detect transcriptional differences.
We performed GSVA for Hallmark cancer gene sets (Figure {@fig:Fig5}B) and quantified immune cell fractions using quanTIseq (Figure {@fig:Fig5}C and Figure {@fig:S6}E), results from which recapitulated previously-described tumor biology. For example, HGG, DMG, MB, and ATRT tumors are known to upregulate MYC [@doi:10.3390/genes8040107] which in turn activates E2F and S phase [@pubmed:11511364]. Indeed, we detected significant (Bonferroni-corrected p < 0.05) upregulation of MYC and E2F targets, as well as G2M (cell cycle phase following S phase) in MBs, ATRTs, and HGGs compared to several other cancer groups. In contrast, LGGs showed significant downregulation (Bonferroni-corrected p < 0.05, multiple cancer group comparisons) of these pathways. Schwannomas and neurofibromas, which have a documented inflammatory immune microenvironment of T and B lymphocytes as well as tumor-associated macrophages (TAMs), are driven by upregulation of cytokines such as IFN$\gamma$, IL-1, and IL-6, and TNF$\alpha$ [@doi:10.1093/noajnl/vdaa023]. Indeed, we observed significant upregulation of these cytokines in GSVA hallmark pathways (Bonferroni-corrected p < 0.05, multiple cancer group comparisons) (Figure {@fig:Fig5}B) and found immune cell types dominated by monocytes in these tumors (Figure {@fig:Fig5}C). We also observed significant upregulation of pro-inflammatory cytokines IFN$\alpha$ and IFN$\gamma$ in both LGGs and craniopharyngiomas when compared to either medulloblastoma or ependymomas (Bonferroni-corrected p < 0.05) (Figure {@fig:Fig5}B). Together, these results support previous proteogenomic findings that aggressive medulloblastomas and ependymomas have lower immune infiltration compared to BRAF-driven LGGs and craniopharyngiomas [@doi:10.1016/j.cell.2020.10.044].
Although CD8+ T-cell infiltration across all cancer groups was quite low (Figure {@fig:Fig5}C), we observed signal in specific cancer molecular subtypes (Groups 3 and 4 medulloblastoma) as well as outlier tumors (BRAF-driven LGG, BRAF-driven and wildtype ganglioglioma, and CNS embryonal NOS; Figure {@fig:S6}E) Surprisingly, the classically immunologically-cold HGGs and DMGs [@doi:10.1186/s40478-018-0553-x; @doi:10.1093/brain/awab155] contained higher overall fractions of immune cells, where monocytes, dendritic cells, and NK cells were the most prevalent (Figure {@fig:Fig5}C). Thus, we suspect that quanTIseq might actually have captured microglia within these immune cell fractions.
While we did not detect notable prognostic effects of immune cell infiltration on overall survival in HGGs or DMGs, we found that high levels of macrophage M1 and monocytes were associated with poorer overall survival (monocyte HR = 2.1e18, 95% CI = 3.80e5 - 1.2e31, p = 0.005, multivariate Cox) in medulloblastomas (Figure {@fig:Fig5}D). We further reproduced previous findings (Figure {@fig:Fig5}E) that medulloblastomas typically have low expression of CD274 (PD-L1) [@doi:10.18632/oncotarget.24951]. However, we also found that higher expression of CD274 was significantly associated with improved overall prognosis for medulloblastoma tumors, although with a marginal effect size (HR = 0.0012, 95% CI = 7.5e−06 - 0.18, p = 0.008, multivariate Cox) (Figure {@fig:Fig5}D). This result may be explained by the higher expression of CD274 observed in WNT subtype tumors by us and others [@doi:10.1080/2162402X.2018.1462430], as this diagnosis carries the best prognosis of all medulloblastoma subgroups (Figure {@fig:Fig5}E).
Finally, we asked whether any subtypes might have a high ratio CD8+ to CD4+ T cells, a metric which has been associated with better immunotherapy response and prognosis following PD-L1 inhibition in non-small cell lung cancer or adoptive T cell therapy in multiple stage III or IV cancers [@doi:10.1136/jitc-2021-004012; @doi:10.4236/jct.2013.48164]. While adamantinomatous craniopharyngiomas and Group 3 and Group 4 medulloblastomas had the highest CD8+ to CD4+ T cell ratios (Figure {@fig:S6}F), very few tumors had ratios greater than 1, highlighting an urgent need to identify novel therapeutics for pediatric brain tumors with poor prognosis. To explore the potential influence of tumor purity, selected transcriptomic analyses were repeated using samples with tumor purities at or above the median tumor purity of their cancer group (see STAR Methods). The analyses using all stranded samples were broadly consistent (Figure {@fig:S7}D-I) with those using samples with high tumor purity.
 {#fig:Fig5 width="7in"}
{#fig:Fig5 width="7in"}