
-
python3
-
DeepTools
-
matplotlib
-
pybedtools
-
pandas
-
subprocess
-
argparse
-
scipy
-
seaborn
-
sklearn
This tool was written using python. Please make sure all the required packages are successfully installed and set them to the environment path. Then it can be downloaded and run directly.
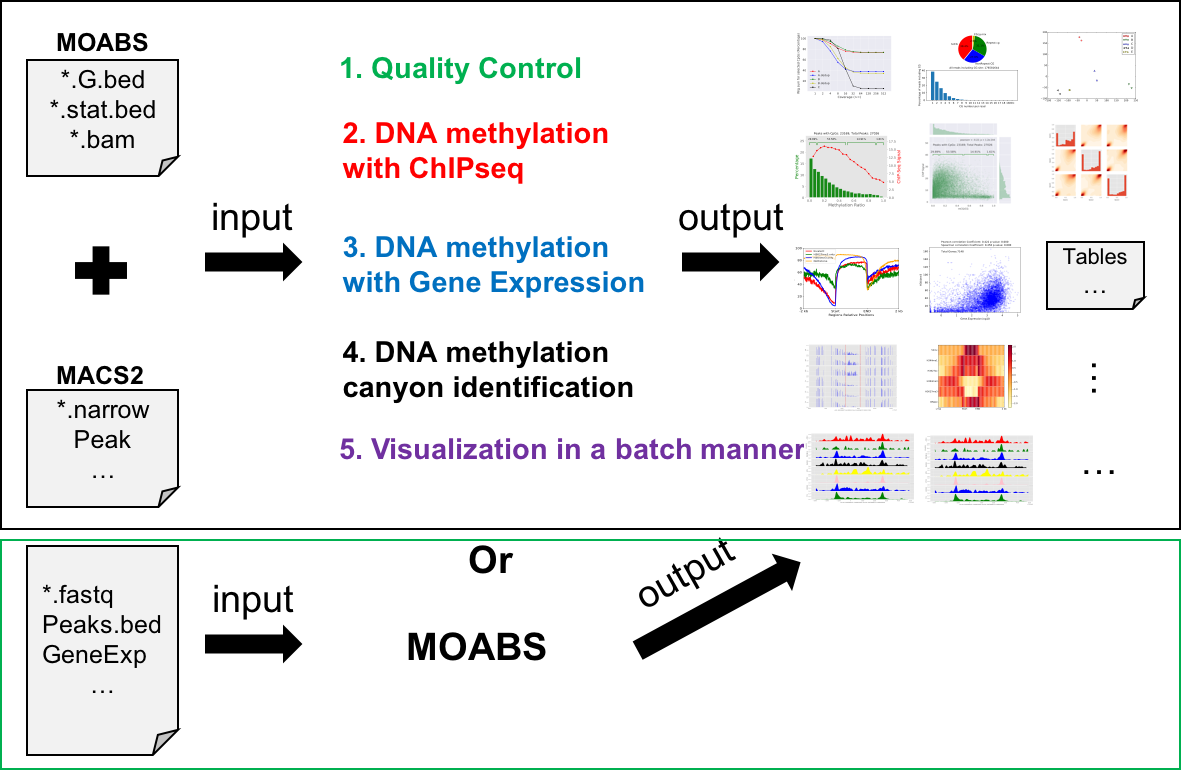
usage: mmint [-h]
{multibw2Bed,multiTracks,multiTracksmeth,CGratio,multibw2multibed,horizonalHeatmap,UMRsExplain,HistonePeaks,HistoneGenes,UMRsCompare,annotate,cluster,DepthVSReadnum,mcor,mdepth,volcano,curveDualYaxis,scatter3d,pca,BedvsExpression,meth2chip}
...
positional arguments:
{multibw2Bed,multiTracks,multiTracksmeth,CGratio,multibw2multibed,horizonalHeatmap,UMRsExplain,HistonePeaks,HistoneGenes,UMRsCompare,annotate,cluster,DepthVSReadnum,mcor,mdepth,volcano,curveDualYaxis,scatter3d,pca,BedvsExpression,meth2chip}
mdepth This script will plot the Mean CpG methylation ratio
and number of CpG sites under a series depth for
multiple samples. The input file is the output file
(*stat.txt) from mcall (MOABS).
pca This script take methylation ratio matrix for multiple
samples as input output a PCA plot.
mcor This script will generate the correlation matrix for
all the samples and some basic statistics. It will
also generate a plot with diagnol as histgram of
methylation ratio for each sample; offdiagnal as
pairwise density for methylation ratio.
meth2chip This script will plot the average ChIP-seq signals at
certain methylation ratio range; the input files are
methylation ratio file (*.G.bed from mcall MOABS) and
the ChIP-seq signals intensity file.
multibw2Bed This script generate the curve plot with multi-bigwig
singal files against interested regions (bed file).
Input files are multiple bigwig files and one bed (or
multiple bed files) file.
multibw2multibed This script generate the curve plot with multi-bigwig
singal files against interested regions (bed file).
Input files are multiple bigwig files and one bed (or
multiple bed files) file.
curveDualYaxis This script generate a two Yaxis (One for mCG/CG; the
other is for ChIP-seq or other datatype) curve plot.
This plot will show how methylation and other data
type distribution on interested regions.
horizonalHeatmap This script will use bigwig and bed files as input and
plot multiple types of signals distribution on
interested locations (up/dnstream xxx bp) as a
horizonal heatmap.
multiTracks This script will use bigwig and bed files as input and
output pictures of multi-bigwig signals for each loci
in the bedfiles. (similar with UCSC visualization
tracks but can generated many automatically)
annotate annotate the bed file
multiTracksmeth help
volcano Volcano plot for bedfile
cluster This script take methylation ratio matrix for multiplt
samples as input to show the cluster result between
samples.
DepthVSReadnum This script will draw figure about depth and reads'
number.
BedvsExpression Bed file versus expression profile
CGratio CG containing reads ratio
HistoneGenes Classification of genes based on two histone markers
HistonePeaks Classification of Peaks based on two histone markers
UMRsCompare Compare UMRs length change between two conditions
UMRsExplain UMRs Explained/overlapped with histones
scatter3d plot 3d scatter with 2d projection
optional arguments:
-h, --help show this help message and exit
- mdepth
This script will plot the Mean CpG methylation ratio and the number of CpG sites under a series depths for multiple samples. The input file is the output file (*stat.txt) from mcall (MOABS).
Example Command:
mmint mdepth -m DE_stat.txt GT_stat.txt FG_stat.txt PE_stat.txt -l DE GT FG PE -o mdepth.pdf
- DepthvsReadsNum
This script will plot the wig sum percentage (Y-axis) (total coverage for certain CpGs/total coverage for all CpGs) at depth >=x (x=1,2,4,8,16,32,64,128,256) (X-axis) to detect if there are high duplication level or sequence bias. If the sequence is randomly sequenced with no bias and no duplication, the total reads number should decrease to less than 10% with the depth increase to certain number (if the read length is 75 bp, the maximum coverage for one CpG supported by non-duplicate reads should be 75); if there is bias or high duplication level, the curve should be a flat line start at certain depth (eg. 100) with percentage of reads >= 20% (see the example).
input file can be the ouput file (*.G.bed) from MOABS (mcall) or a file with at least 6 columns:
chr start end ratio total-C total-mC
Example Command:
mmint DepthvsReadsnum -m a.G.bed b.G.bed c.G.bed -l A B C -o depthvsReadsNum
- CGratio
This script will calculate the distributions of reads based on if it include CGs and where the reads from (Repeat regions;Non-repeat regions). It will generate one piechart and one histogram. The piechart can tell you the distributions of reads based on if the reads include CG and if the reads are from repeat regions; the histogram can tell you the ratio of reads that include how many CGs in one read.
- PCA and cluster
This script take methylation ratio matrix for multiple samples and the bed file for intereseted regions as input and output a PCA/cluster plot based on mCG/CG on certain regions (e.g. promoter, CpG Island ...) with certain coverage (e.g. >=10).
Example Command:
A.G.bed is the output file from MAOBS (mcall) or it can be bed files with at least five columns:
chr start end methRatio depth
mmint pca -i A.G.bed B.G.bed C.G.bed -n 3 -r 1 -N A B C -b cpgisland.bed -c 10 -o meth.PCA
mmint cluster -i A.G.bed B.G.bed C.G.bed -N A B C -o cluster -l average
- mCor
This script will generate the correlation matrix for all the samples under certain coverage threshold and some basic statistics. It will also generate a plot with diagnol as histgram of methylation ratio for each sample; offdiagnal as pairwise density for methylation ratio.
The input file is the same with PCA;
Example Command:
mmint mcor -m test1.bed test2.bed test3.bed -o mCorr
- Meth2ChIP
This script will plot the average ChIP-seq signals at certain methylation ratio range (5% intervals); We separated the mehtylation level to four categories: UMR(0-0.1),LMR(0.1-0.5),MMR(0.5-0.9),HMR(0.9-1.0); and it will also output the numbers of regions that include the CpGs in your inputfile; the ratios of regions that fall into the four categories, respectively. In addition, it will generate a scatter plot with Xaxis as mCG/CG ratio and Yaxis as ChIPseq intensity. The input files are methylation ratio file (*.G.bed from mcall MOABS) and the ChIP-seq signals intensity file.
Example Command:
mmint meth2chip -m test.G.bed -p ChIP.bed -o meth2ChIP
ChIP-seq signals intensity file:
chr peak.start peak.end intensity
chr1 100113803 100113954 0.219254
chr1 100715377 100715412 0.1062
chr1 1007642 1007662 0.107425
chr1 100816764 100816969 0.35706
chr1 100817693 100817732 0.113485
- multiBw2bed / multiBw2multiBed
This script generate the curve plot with multi-bigwig singal files against interested regions (one bed file or multiple bed files). Input files are multiple bigwig files and one bed (or multiple bed files) file.
Example command:
mmint multiBw2multiBed -bw H3K4me1.meth.bw H3K27me3.meth.bw H3K27ac.meth.bw ATAC.meth.bw -bed H3K4me1peaks.bed H3K27me3peaks.bed H3K27acpeaks.bed ATACpeaks.bed -after 2000 -before 2000 -bs 20 -m 2000 -L H3K4me1 H3K27me3 H3K27ac ATAC -n methOnMultipleRegions.pdf
- curveDualYaxis.py
This script generate a two Yaxis (One for mCG/CG ratio; the other is for ChIP-seq or other datatype) curve plot. This plot will show how methylation and other data type distribution on interested regions.
mmint curveDualYaxis -bw PE.final.meth.bw d8.ATAC.bw -bed d8_peaks.narrowPeak -after 1000 -before 1000 -bs 20 -m 1000 -L mCG/CG ATAC.signal -n PE.meth.ATAC2 -xlab ATACpeaks -ylab1 mCG/CG -ylab2 ATACsignal
- horizontalHeatmap.py
This script will use bigwig and bed files as input and plot multiple types of signals distribution on interested locations (up/dnstream xxx bp) as a horizonal heatmap.
Example command:
mmint -bw H3K4me1.bw H3K27me3.bw H3K27ac.bw ATAC.bw -bed interested.bed -after 2000 -before 2000 -bs 20 -m 2000 -L H3K4me1 H3K27me3 H3K27ac ATAC -n horizonal.heatmap.pdf
- multiTracks.py/multiTracks.meth.py
This script will use bigwig and bed files as input and output pictures of multi-bigwig signals for each loci in the bedfiles. (similar with UCSC visualization tracks but can generated many automatically) The multiTracks.meth.py is taking methylation bw files as input and output the up/dn 5000 bp of the intereseted regions;with red dashed lines as the boundaries of the interested regions.
Example command:
mmint multiTracks -bs 100 -f regions.txt -b H3K4me3_m24.merge.bw H3K27me3_m24.merge.bw KO-K4m3.norm.bw KO-K27m3.norm.bw -labels m_H3K4me3 m_H3K4me3 KO_H3K4me3 KO_H3K27me3 -c r g b black yellow
regions.txt:
chr4:82878750:82888151 geneName1
chr14:56255857:56265260 geneName2
chr7:44809255:44818658 geneName3
chr7:45915022:45924426 geneName4
chr8:85068756:85078161 geneName5
- volcano
This script will draw volcano plot about bed files
Example command:
mmint volcano -f dmrtest.bed -p 5 -o test
dmrtest.bed:
chr1 start end methylation_difference p_value
Fields seperate by tab.
- BedvsExpression
This script will calculate the correlation between mCG/CG ratio on certain regions with associated genes expression. e.g. if you want to investigate how promoter mCG/CG correlate with associated gene expression, you just need to input methylation ratio, the gene expression file (two columns: GeneName\tFPKM) and the TSS bed file. It will generate a scatter plot to demonstrate the correlation.
Example command:
mmint bedvsexpression -m methRatio.bed -r hg19 -u 1000 -d 1000 -R gene.exp -o methvsExp -ylab Genes.Exp.log10 -xlab mCG/CG
- HistoneGenes
This script will classify genes based on Histone peaks, so you can investigate how these two histones interact with DNA methylation on different groups of genes classified by these two histones (eg. bivalend promoter (H3K4me3 and H3K27me3); active/inactive enhancers (H3K4me1 and H3K27ac)). It takes two different histones peaks, mCG/CG ratio bigwig file as input and output four genes groups (only histone A; only histone B; both histone A and B; No Histones) based on calculating the overlaps among two histone peaks and regions around TSS. It can give you four curves of mCG/CG ratio for the four classies along gene regions.
mmint HistoneGenes -tss mm10.tss.bed -gene genes.bed -b1 histone1.peaks.bed -b2 histone2.peaks.bed -o1 -o test1.gz test2.gz test3.gz test4.gz -bw methRatio.bw -m 2000 -after 2000 -before 2000 -bs 20 -L Overlap histone1Only histone2Only NoHistone -n HistoneGenes.pdf
The format of mm10.tss.bed:
The format of mm10.GENE.bed (tab separated and should sorted by using sort -k4,4b):
chr7 74817817 74819817 0610006L08Rik chr12 85814447 85816447 0610007P14Rik chr11 51684385 51686385 0610009B22Rik chr2 26444695 26446695 0610009E02Rik chr11 120347677 120349677 0610009L18Rik
- HistonePeaks
This script is similar with HistoneGenes. It will classify the histone peaks based on the overlap between thw different histones peaks. And, then it can generate the mCG/CG curve on these four histone peaks groups (only histone A; only histone B; both histone A and B; No Histones).
mmint HistonePeaks -b1 histone1.peaks.bed -b2 histone2.peaks.bed -o1 Overlap.bed histone1Only.bed histone2Only.bed NoHistone.bed -o test1.gz test2.gz test3.gz test4.gz -bw methRatio.bw -after 2000 -before 2000 -bs 20 -L Overlap histone1Only histone2Only NoHistone -n HistoneOverlap.pdf
-
DNA methylation Canyon
-
UMRsCompare
mmint UMRsCompare -1 N.UMRs -2 T.UMRs -o UMRsCompare
UMRs/canyons file format:
chr10 5822782 5826476 hmr25_0.0394569 94 +
chr10 7390304 7393815 hmr42_0.0266945 183 +
chr10 7953879 7957683 hmr57_0.00891266 68 +
chr10 9898943 9902518 hmr78_0.0190414 171 +
chr10 13964044 13968496 hmr123_0.00123365 193 +
- UMRsExplain
mmint UMRsExplain -i N.UMRs -1 histone1.bed -2 histone2.bed -o UMRsExpained
- scatter3d
mmint scatter3d -DMRs DMRs.bed -histRs histone.fold.bed -o scatter3d.pdf
Jia Li: [email protected] / [email protected]; Yue Yin: [email protected]