Testing gatk4 module on Atlas HPC
Reuse dataset from GATK nextflow pipeline (GitHub:HuffordLab/Maize_WGS_Build)
wget https://iastate.box.com/shared/static/wt85l6s4nw4kycm2bo0gpgjq752osatu.gz
tar -xf wt85l6s4nw4kycm2bo0gpgjq752osatu.gz
ls -1 test-data/
#> fastq # <= folder of paired reads
#> read-group.txt
#> ref # <= folder containing one genome reference
(1) Load gatk module
salloc -A isu_gif_vrsc
module load singularity
module use /project/users/jim.coyle/modulefiles
module load gatk
Turns out gatk is a function:
which gatk
gatk is a function
gatk ()
{
singularity exec /project/users/jim.coyle/containers/gatk/4.2.0.0//gatk-4.2.0.0.sif gatk "$@"
}
Bind to current working directory or it will end up with strange pathing errors (can't find input files, any output files tend to be sent to home directory instead of current directory)
gatk2 ()
{
singularity exec --bind $PWD /project/users/jim.coyle/containers/gatk/4.2.0.0//gatk-4.2.0.0.sif gatk "$@"
}
(2) Print help message
gatk2 --list
#> Using GATK jar /gatk/gatk-package-4.2.0.0-local.jar
#> Running:
#> java -Dsamjdk.use_async_io_read_samtools=false -Dsamjdk.use_async_io_write_samtools=true -Dsamjdk.use_async_io_write_tribble=false -Dsamjdk.compression_level=2 -jar /gatk/gatk-package-4.2.0.0-local.jar --help
#> USAGE: <program name> [-h]
#>
#> Available Programs:
#> --------------------------------------------------------------------------------------
#> Base Calling: Tools that process sequencing machine data, e.g. Illumina base calls, and detect sequencing level attributes, e.g. adapters
#> CheckIlluminaDirectory (Picard) Asserts the validity for specified Illumina basecalling data.
#> CollectIlluminaBasecallingMetrics (Picard) Collects Illumina Basecalling metrics for a sequencing run.
#> CollectIlluminaLaneMetrics (Picard) Collects Illumina lane metrics for the given BaseCalling analysis directory.
#> ExtractIlluminaBarcodes (Picard) Tool determines the barcode for each read in an Illumina lane.
#> IlluminaBasecallsToFastq (Picard) Generate FASTQ file(s) from Illumina basecall read data.
# ...
# === Input
REF=00_Raw-Data/test-data/ref/b73_chr1_150000001-151000000.fasta
R1=00_Raw-Data/test-data/fastq/BioSample01_R1.fastq.gz
R2=00_Raw-Data/test-data/fastq/BioSample01_R2.fastq.gz
READNAME=BioSample01
The steps are slighly modified from the GATK workbook
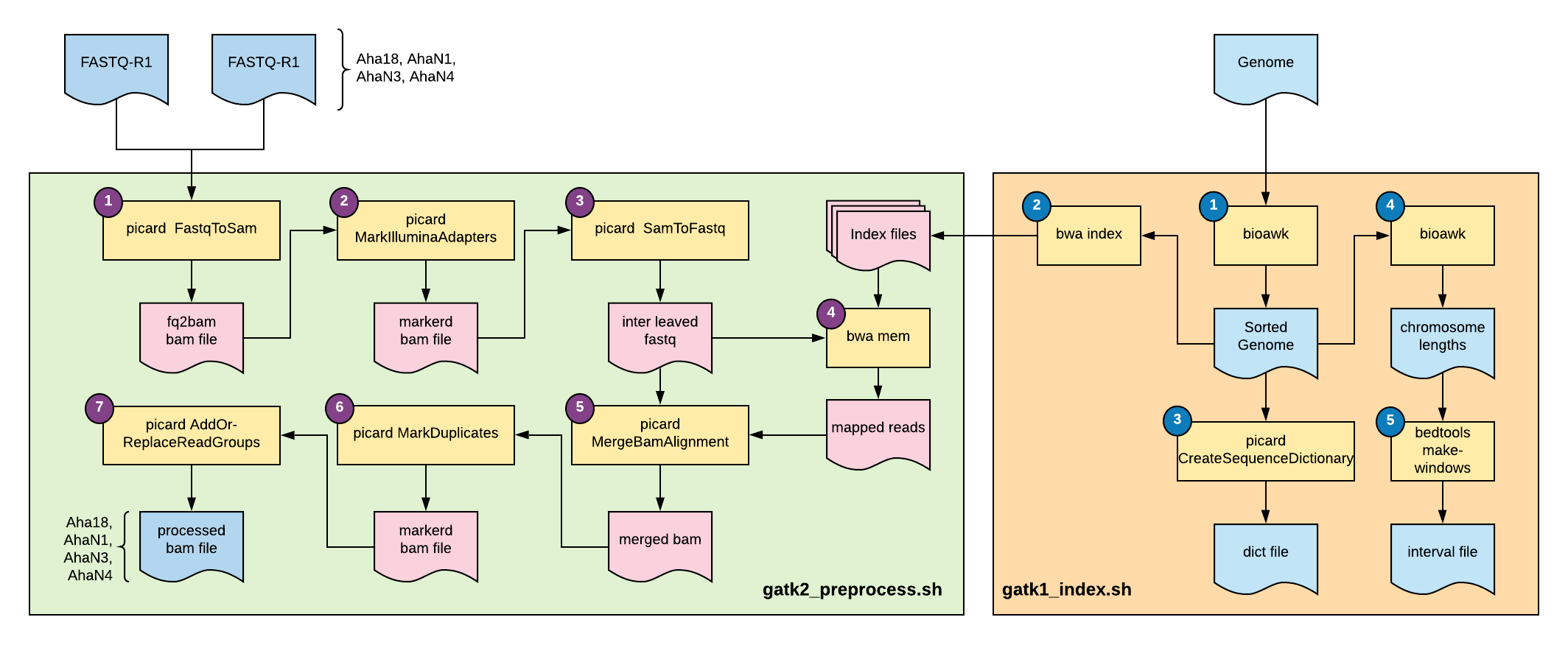
(1) FastqToSam
gatk2 FastqToSam \
--FASTQ $R1 \
--FASTQ2 $R2 \
--OUTPUT ${READNAME}.bam \
--READ_GROUP_NAME ${READNAME} \
--SAMPLE_NAME ${READNAME}_name \
--LIBRARY_NAME ${READNAME}_lib \
--PLATFORM ILLUMINA \
--SEQUENCING_CENTER ISU
(2) MarkIlluminaAdapters
gatk2 MarkIlluminaAdapters \
--INPUT ${READNAME}.bam \
--OUTPUT ${READNAME}_marked.bam \
--METRICS ${READNAME}_marked_metrics.txt
(3) SamToFastq
gatk2 SamToFastq \
--INPUT ${READNAME}_marked.bam \
--FASTQ ${READNAME}_newR1.fq \
--SECOND_END_FASTQ ${READNAME}_newR2.fq \
--CLIPPING_ATTRIBUTE XT \
--CLIPPING_ACTION 2 \
--INTERLEAVE true \
--INCLUDE_NON_PF_READS true
# --FASTQ ${READNAME}_interleaved.fq
(4) bwa index
Needed to load bwa or bwa-mem2 separately, was not included in gatk module. (We should include it, along with samtools)
bwa-mem2 index $REF
(5) bwa mem
PROC=2
bwa-mem2 mem -t $PROC $REF ${READNAME}_newR1.fq ${READNAME}_newR2.fq |\
samtools view --threads $PROC -bS - > ${READNAME}_aln.bam
(6) CreateSequenceDictionary
gatk2 CreateSequenceDictionary \
-R $REF \
-O ${REF}.dict # Actually will need to drop off fasta extension
(7) MergeBamAlignment
gatk2 MergeBamAlignment \
--REFERENCE_SEQUENCE $REF \
--UNMAPPED_BAM ${READNAME}.bam \
--ALIGNED_BAM ${READNAME}_aln.bam \
--OUTPUT ${READNAME}_merged.bam \
--CREATE_INDEX true \
--ADD_MATE_CIGAR true \
--CLIP_ADAPTERS false \
--CLIP_OVERLAPPING_READS true \
--INCLUDE_SECONDARY_ALIGNMENTS true \
--MAX_INSERTIONS_OR_DELETIONS -1 \
--PRIMARY_ALIGNMENT_STRATEGY MostDistant \
--ATTRIBUTES_TO_RETAIN XS
(8) MarkDuplicates
gatk2 MarkDuplicates \ #<=hmm we seemed to skip this step in our nextflow pipeline
...
(9) AddOr-ReplaceReadGroups
...
(10?) Make Windows
samtools faidx $REF
awk -F'\t' '{print $1"\t"$2}' ${REF}.fai > genome_length.txt
bedtools makewindows -w 100000 -g genome_length.txt |\
awk '{print $1"\t"$2+1"\t"$3}' |\
sed 's/\t/:/1' |\
sed 's/\t/-/g' > ${REF}_coords.bed
head ${REF}_coords.bed
#> chr1:1-100000
#> chr1:100001-200000
#> chr1:200001-300000
(11) GATK HaplotypeCaller (finally!)
gatk2 HaplotypeCaller \
-R $REF \
-I ${READNAME}_merged.bam \
-L chr1:1-100000 \
-O chr1_1-100000.vcf
# === gatk reference command below, might still need --java-options
# gatk_app --java-options \"-Xmx80g -XX:+UseParallelGC\" HaplotypeCaller -R $genome_fasta -I \$BAMFILES -L $window --output ${window.replace(':','_')}.vcf
Usually running for several windows (in parallel), so would need to merge results... skipping for now.
(12) Sort and calc DP value
gatk2 SortVcf \
--INPUT chr1_1-100000.vcf \
--SEQUENCE_DICTIONARY ${REF}.dict \
--CREATE_INDEX true \
--OUTPUT chr1_1-100000_sorted.vcf
grep -v "^#" chr1_1-100000_sorted.vcf | cut -f 8 | grep -oe ";DP=.*" | cut -f 2 -d ';' | cut -f 2 -d "=" > dp.txt
# module load datamash # Geh, why doesn't this work? Hmm, this might not be an Atlas module...
# Okay for now installed in a conda env: "conda install -c bioconda datamash"
cat dp.txt | datamash mean 1 sstdev 1 > dp.stats
cat dp.stats | awk '{print $1+5*$2}'
#> 187.433
(13) gatk VariantFiltration
gatk2 VariantFiltration \
--reference ${REF} \
--sequence-dictionary ${REF}.dict \
--variant chr1_1-100000_sorted.vcf \
--filter-expression "QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0 || DP > 187.433" \
--filter-name FAIL \
--output chr1_1-100000_sorted_marked.vcf
(14) Keep only passing
cat chr1_1-100000_sorted_marked.vcf | grep "^#" > chr1_1-100000_sorted_marked_snp-only.pass-only.vcf
cat chr1_1-100000_sorted_marked.vcf | grep -v "^#" | awk '$7=="PASS"' >> chr1_1-100000_sorted_marked_snp-only.pass-only.vcf
nextflow run 04_GATK.nf \
--genome "00_Raw-Data/test-data/ref/*.fasta" \
--reads "00_Raw-Data/test-data/fastq/*_{R1,R2}.fastq.gz" \
-resume \
-with-singularity gatk.sif \
-with-timeline "timeline_report.html"
N E X T F L O W ~ version 20.10.0
Launching `04_GATK.nf` [voluminous_hodgkin] - revision: 89ee103d4e
executor > slurm (155)
[a8/0b122d] process > bwamem2_index (b73_chr1_150... [100%] 1 of 1 ✔
[eb/749bc1] process > bwamem2_mem (BioSample19) [100%] 27 of 27 ✔
[c2/ba080c] process > FastqToSam (BioSample12) [100%] 27 of 27 ✔
[1f/e64577] process > MarkIlluminaAdapters (15_Bi... [100%] 27 of 27 ✔
[af/837898] process > SamToFastq (15_BioSample12_... [100%] 27 of 27 ✔
[5a/7467d4] process > CreateSequenceDictionary (b... [100%] 1 of 1 ✔
[35/fd20b7] process > samtools_faidx (b73_chr1_15... [100%] 1 of 1 ✔
[37/72890f] process > MergeBamAlignment (19_BioSa... [100%] 27 of 27 ✔
[96/6dffa1] process > makewindows (b73_chr1_15000... [100%] 1 of 1 ✔
[ba/becb4e] process > gatk_HaplotypeCaller (chr1:... [100%] 10 of 10 ✔
[0e/387bc4] process > merge_vcf (merging) [100%] 1 of 1 ✔
[47/6bba6c] process > vcftools_snp_only (first-ro... [100%] 1 of 1 ✔
[37/94898c] process > SortVcf (first-round_merged... [100%] 1 of 1 ✔
[6d/54dfab] process > calc_DPvalue (first-round_m... [100%] 1 of 1 ✔
[52/52f702] process > VariantFiltration (first-ro... [100%] 1 of 1 ✔
[81/26ce16] process > keep_only_pass (first-round... [100%] 1 of 1 ✔
Completed at: 06-Apr-2021 17:27:13
Duration : 47m 48s
CPU hours : 6.0
Succeeded : 155
- timeline_report.html
- Compare Nextflow processes with WDL processes, notice similarity in naming - gatk4-rna-best-practices.wdl