anvi'o v7, "hope"
We are happy to announce anvi'o v7 with the code name, "hope"!
After more than 3,000 changes that introduced about 35,000 new lines of code, this stable release of anvi'o represents one of the largest leaps forward in the history of the platform that introduces many new features, performance improvements, and fixes for known bugs.
This page intends to give you a summary of some of the notable changes that come with hope.

The code name recognizes Hope E. Hopps as a tribute to all laboratory technicians whose contributions have often been poorly recognized in science. This is despite the fact that technicians not only ensure accuracy, efficiency, and reproducibility in any laboratory, but also push the boundaries of science as much as any other member of their groups, if not more in many cases. Hopps was a specialist in infectious diseases and in 1966 she developed, together with Harry M. Meyer and Paul J. Parkman, a highly effective vaccine for rubella, a viral infection which caused more than 30,000 stillbirths in the United States alone between 1962 and 1965. Despite her role in the vaccine development, in a historical photograph by the NIH that portrays the rubella vaccine development team, Hopps was only identified as "Female Lab Technician" until recently, even though the caption of the same photograph explicitly named Meyer and Parkman. The unfair treatment of laboratory technicians remains to be commonplace in today's science. In fact, "not more than a technician's job" can serve as an argument for professors when they wish to refuse the recognition of one's contributions to science. We can't ignore the significant progress we have made as a community during the past few years. But while we continue working on increasing the diversity, equity, and inclusion in science, we must also recognize and face the implicit and explicit biases against those in science who are not PIs, post-docs, or graduate students.
Disclosures: The code name was a suggestion by Alon Shaiber, a Genomics Data Scientist at Weill Cornell Medicine. The release notes were written by Meren and proofread by Iva Veseli. Alon, Meren, and Iva are among the developers of anvi'o.
New help pages for anvi'o programs and artifacts
As anvi'o developers, we always knew the critical importance of providing our users with extensive tutorials so they can find their way through their data themselves. However, as anvi'o matured, the number of anvi'o programs and artifacts increased dramatically. This created a bottleneck since every anvi'o tutorial assumed that our users knew about the common concepts in anvi'o (such as 'the profile database' or 'a collection') or common anvi'o programs (such as 'anvi-profile' or 'anvi-interactive'). Solving this fundamental problem required us to think of an entirely new technical approach to our documentation that is now in place.
We have now implemented a system (#1425) that makes two things possible:
- (1) For anvi'o developers, a means to quickly describe their contributions without leaving the environment where they write code (for instance, here is the description of 'collection' in the codebase),
- (2) For anvi'o users, a means to be able to see that information on a web page where all anvi'o programs and concepts are interconnected (for instance, here is the description of 'collection' on the web page).
This way, anvi'o could accumulate information from its developers without burdening them and present it to its users in a way where self-learning is possible. However, there was one significant problem: retrospectively describing all the things that have already been implemented in the codebase. Enter Jessica Pan (@Jessica-Pan), an undergraduate student at the MIT. Jessica took the responsibility of describing existing anvi'o programs and artifacts a few months ago and with the guidance of other anvi'o developers, Jessica was able to populate this technical framework with her words and descriptions (#1470), which added more than 200 files and tens of thousands of words of documentation to the codebase.
With this release, we are happy to also release the first outputs of this documentation project here, with the hope that it will make your life with anvi'o a little easier going forward:
Perhaps it will not be a surprise to the long-term anvi'o users that this documentation system is also connected to our command-line programs. Thanks to this, they will be able to offer you more useful help menu outputs. For instance, if you were to type anvi-interactive --help in your terminal in v7, you would see the following section at the end of the help menu, so you can click on the link to go to the online description of the program and browse through examples and artifacts associated with it:

If you visit the help pages you will see there are 'edit' links under every file. It is our way of inviting the rest of the community to contribute to these pages with their own experiences with anvi'o tools. If you have ideas to make it better, come to our Slack channel for a discussion, or file a GitHub issue. We are all ears.
Significant performance improvements
This version will likely be remembered for significant performance improvements by multiple heroes, including Evan Kiefl (@ekiefl), Iva Veseli (@ivagljiva), and Ryan Moore (@mooreryan). Here is a glimpse of what happened in v7 compared to v6:
-
Profiling BAM files is one of the most critical steps in anvi'o and the program anvi-profile has been a nightmare for memory and processing time. Thanks to @ekiefl's significant improvements that influenced the runtime and memory requirements of the profiling step (#1362, #1339),
anvi-profileis now ~17 times faster. -
One of the first steps in any anvi'o workflow is turning boring FASTA files into talented contigs databases that can be used by many anvi'o programs. Yet, the program anvi-gen-contigs-database has not been multithreaded, which has been a significant performance bottleneck before (#1344, #1431). Responding to our plea on Twitter, @mooreryan has made remarkable contributions to the anvi'o codebase (while wrapping up his PhD, that is) (#1437, #1468, and #1445). As a result, anvi-gen-contigs-database is now multithreaded, at least two times faster than before, and can take advantage of your fancy clusters to be even more efficient.
-
After making our users who care about the quality of their MAGs go through so much pain and suffering for years, anvi-refine in this release is ~13 times faster after significant improvements in its memory requirements (#1455, #1458), thanks to help from Xabier Vázquez-Campos (@xvazquezc).
-
Our dealings with HMMs also benefited from major performance improvements. Thanks to @ivagljiva's efforts (#1413), the two frequently used programs, anvi-run-hmms and anvi-run-pfams that rely on HMMER, will perform HMM annotations ~3x faster.
An integrated subsystem for metabolic reconstruction
We are also thrilled to announce that starting with this release, anvi'o includes a new suite of programs for predicting metabolic capabilities for genomic and metagenomic data, thanks to @ivagljiva's extensive work that started with #1413. The new programs in this release rely on the extensive set of resources in the Kyoto Encyclopedia of Genes and Genomes (KEGG) for gene annotation and metabolism estimation, although in future releases we will expand source resources for metabolic reconstruction.
As a part of this subsystem, this release introduces a new database, the anvi'o MODULES.db, which is generated from parsed KEGG data files (such as KOfam HMM profiles and text-based descriptions of KEGG MODULE) and used by subsequent programs detailed below for easy, organized access to metabolic data. A version tracking system ensures metabolism estimation is run using the same MODULES.db that was used to annotate a given CONTIGS.db. Here are the key programs for the anvi'o subsystem for metabolism:
-
The program anvi-setup-kegg-kofams generates a
MODULES.dband stores it on your server or the local computer. -
The program anvi-run-kegg-kofams annotates genes in a given anvi'o contigs database with KEGG Orthology (KO) numbers (via hits to the KEGG KOfam database). This program is included in the anvi'o snakemake workflows Alon Shaiber (@ShaiberAlon) had introduced, which enables bulk annotation of several contigs databases with a single command.
-
The program anvi-estimate-metabolism predicts metabolic potential in a given set of sequences by integrating KO annotations with KEGG MODULE information. These estimates are integrated into anvi'o in various ways and can be summarized in flat text files. In addition to contigs databases, and optionally profile databases and collections, the program accepts internal, external, or metagenomes files as input. The program is able to work with a variety of output options.
We look forward to the input from the community to offer improved and integrated metabolic insights into microbial genomes and metagenomes.
New tools for Transfer RNA biology
One of the most significant advances in this release include the new tools developed by Samuel Miller (@semiller10) for the study of Transfer RNA transcripts that are generated by tRNA-seq, a sequencing protocol that is developed by the members of Tao Pan's group at the University of Chicago. This sequencing strategy aims to offer high-resolution insights into the translational regulation of cells by revealing changes in the abundance and chemical modifications of tRNA transcript across environmental conditions. While the sequencing strategy makes accessible tRNA transcripts from diverse environments, the extremely complex data generated by this strategy requires completely new computational approaches not only to characterize tRNA sequences with their structural properties but also to resolve chemical modification fractions and their taxonomy. We hope that the more than 10,000 lines of code @semiller10 has created behind anvi-trnaseq and anvi-convert-trnaseq-database (primarily through #1509 and #1615) and their cousin programs anvi-scan-trnas and anvi-estimate-trna-taxonomy, will set the stage for new horizons that can bring more RNA biology into the anvi'o ecosystem in an integrated fashion.
Ability to profile INDELs in mapping results
Anvi'o offers a powerful and comprehensive framework to enable in-depth investigations of microbial population genetics. Yet, these insights have so far been limited to single-nucleotide variants, single-codon variants, and single-amino acid variants as reported and/or used by anvi-gen-variability-profile, anvi-display-structure, and anvi-gen-fixation-index-matrix or as displayed in varioius interactive interfaces.
This version comes with a completely redesigned anvi-profile, which is now able to characterize INDELs in read recruitment results thanks to @ekiefl's additions to the codebase especially in #1394. The ability to characterize INDELs pushes the boundaries of our ability to make sense of microbial population genetics through metagenomes, and we hope you will find many gems in your data, as well.
Currently, anvi'o reports INDEL tables that are ready to go into R or any other statistical analysis or visualization environment that you can obtain via the program anvi-export-table with --table indels:

To benefit from this feature in your existing projects, you will need to re-profile your BAM files ... which should be easy-peasy if you have been using anvi'o snakemake workflows ;)
Improvements in the interactive interface
The power of anvi'o in the command line is complemented by its interactive interfaces, which also benefit from numerous new features in this release. But perhaps the most critical improvements were those contributed by Isaac Fink (@isaacfink21), an undergraduate student at the University of Chicago, who revamped the inspection page (in #1466 and others).
Not only we are now able to visualize single-nucleotide variants better,
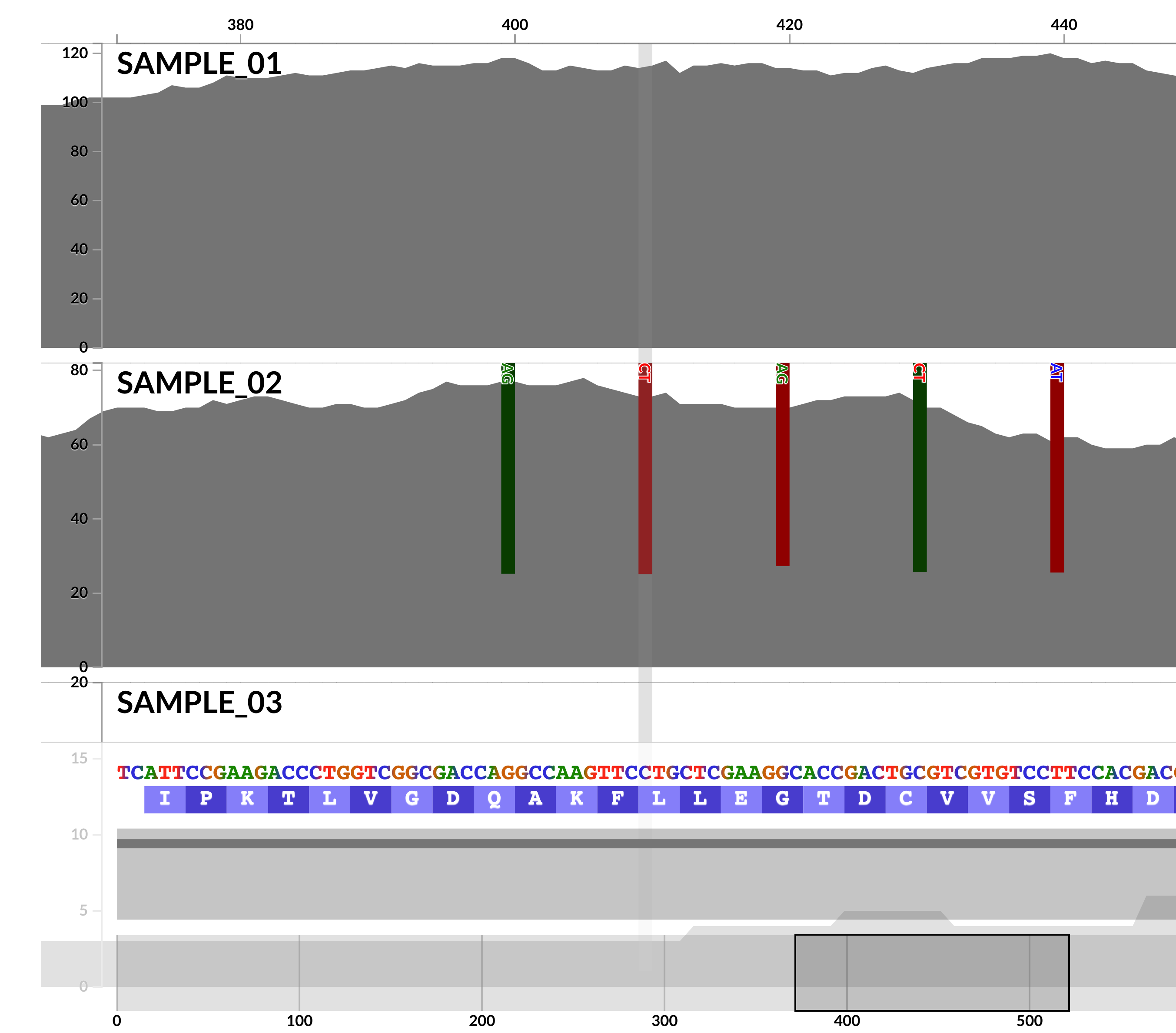
but also the interactive interface is now able to visualize INDELs in (meta)genomic/(meta)transcriptomic read recruitment results,
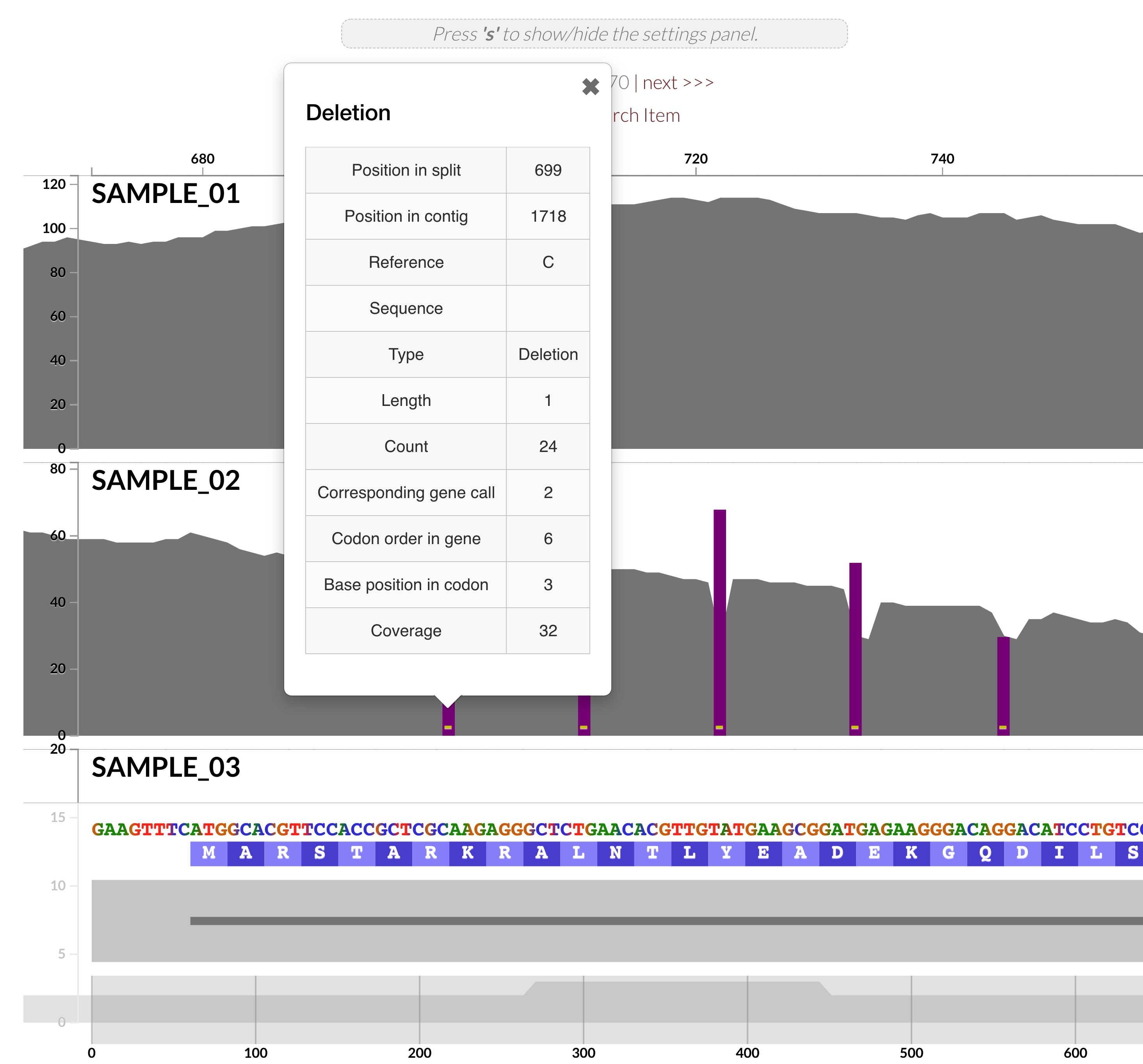
so you can spend EVEN MORE TIME looking at your coverages.
Isaac Fink's revamped inspection page comes with a 'settings' panel, organizing all the features this page offers, which looks like this:

There are other exciting features that will likely make those who use anvi'o for pangenomics and phylogenomics very happy.
Anvi'o interactive interface was not programmed to show support values for phylogenomic trees, which was a long standing item (#1450) in the list of feature requests we had. Matthew Lawrence Klein (@matthewlawrenceklein) who joined our team only a few weeks ago, managed to improve this shortcoming through #1618 thanks to the guidance we received from Tom Delmont. Result? Anvi'o can now visualize branch support values on trees when applicable:


While this is a preliminary implementation of this feature, we are looking forward to the feedback we will receive from the community to improve it.
Another critical shortcoming was the difficulty of selecting multiple items using the interactive interface when there was no tree or dendrogram to guide the selection of objects. This happened especially working with pangenomes, where ordering gene clusters based on their synteny is a common need, yet selecting regions of interest requires clicking on each item individually. @matthewlawrenceklein also addressed this through #1614, and it is now possible to select a range of items in a straightforward fashion:

On top, after selecting regions of interest in a pangenome, it is now possible to take a quick peek at functions gene clusters encode through the interactive interface without having to run anvi-summarize:

Other fancy tools and functionality
-
Ability to estimate per-residue binding frequencies for protein strucutres with anvi-run-interacdome by Evan Kiefl. This enables very highly resolved analyses of environmental variants in conjunction with the structural context of proteins and their ligand binding sites. If this sounds interesting to you, read Evan's journey implementing this feature, see his gargantuan of a pull request at #1472, or read this paper by Shilpa Nadimpalli Kobren and Mona Singh to get yourself familiarized with the goal here.
-
Ability to extract gene loci or operons from any genomic context with anvi-export-locus by Matthew Schechter (@mschecht) and others. By enabling high-throughput recovery of loci of interest across any number of genomes by marking genes with their functional annotations or HMM hits, this strategy makes it possible to ask very specific questions regarding the gene content, evolution, and ecology of genomic operons. You can read Matt's tutorial here, or see is pull request at #1386.
-
Generalization of the functional enrichment analysis in #1500 by Iva Veseli. This statistical approach was initially developed by Amy Willis (@adw96) and was implemented in the anvi'o program
anvi-get-enriched-functions-per-pan-group. As the name suggests, this tool was specific to studying functional enrichment in pangenomes. Thanks to Iva's contribution, the new program anvi-compute-functional-enrichment is now able to work with pangenomes, metabolic pathways, and internal or external genomes to study functional enrichment statistics between distinct groups of entities. -
Integration of the 2020 release of the NCBI's Clusters of Orthologous Groups database to increase the utility of anvi-run-ncbi-cogs for functional annotations (#1570).
And many more.
A list of new anvi'o programs
This release comes with the following programs that were not in the previous stable release: anvi-convert-trnaseq-database, anvi-display-metabolism, anvi-estimate-metabolism, anvi-estimate-trna-taxonomy, anvi-run-interacdome, anvi-run-kegg-kofams, anvi-run-trna-taxonomy, anvi-script-augustus-output-to-external-gene-calls, anvi-script-fix-homopolymer-indels, anvi-script-gen-pseudo-paired-reads-from-fastq, anvi-script-get-primer-matches, anvi-script-pfam-accessions-to-hmms-directory, anvi-script-tabulate, anvi-setup-interacdome, anvi-setup-kegg-kofams, anvi-setup-scg-taxonomy, anvi-setup-trna-taxonomy, anvi-trnaseq.
Anvi'o as a community platform: ✅
We have always imagined anvi'o as a community platform, and we are getting there. Even this very release is a product of voluntary contributions of many members of the anvi'o community, who slowly shape this open-source software ecosystem for integrated multi-omics.
A few weeks ago we published a comment that was authored by those who are mentioned in these release notes and many more who have been supporting anvi'o in many ways to make it more accessible to the community of microbiologists.

Our paper ends with this statement:
(...) As an open-source platform that empowers microbiologists by offering them integrated yet uncharted means to steer through complex ‘omics data, anvi’o welcomes its new users and contributors.
We thank you for your interest in anvi'o and for your patience with it, in advance. We hope that anvi'o will continue to empower you in 2021 so you can find the answers you are looking for in the avalanche of data that surrounds you.
See our up-to-date installation instructions here, which include docker and conda solutions and ways to reach out to the anvi'o community for help if you run into a problem.