Home
iCellR is an interactive R package to work with high-throughput single cell sequencing technologies (i.e scRNA-seq, scVDJ-seq, scATAC-seq, CITE-Seq and Spatial Transcriptomics (ST)).
News (April 2021): Use iCellR version 1.6.4 for scATAC-seq and Spatial Transcriptomics (ST). Use the i.score function for scoring (scoring cells based on gene signatures) methods (i.e. tirosh, mean, sum, gsva, ssgsea, zscore and plage).
News (July 2020): See iCellR version 1.5.5 with new cell cycle analysis for G0, G1S, G2M, M, G1M and S phase, Pseudotime Abstract KNetL map (PAK map) and gene-gene correlations. See below for how to.
News (May 2020): see our dimensionality reduction called KNetL map  (pronounced like "nettle"). KNetL map is capable of zooming and shows a lot more details compared to tSNE and UMAP.
(pronounced like "nettle"). KNetL map is capable of zooming and shows a lot more details compared to tSNE and UMAP.
News (April 2020): see our imputation/coverage correction (CC) and batch alignment (CCCA and CPCA) methods. More databases added for cell type prediction (ImmGen and MCA).
- Tutorial: example 1 code and results (based on KNetL map )
- Tutorial: example 2 code and results (based on CPCA batch alignment and KNetL map )
- Link to a video tutorial for CITE-Seq and scRNA-Seq analysis: Video
- All you need to know about KNetL map: Video
- Link to manual Manual and Comprehensive R Archive Network (CRAN).
iCellR Viewer (web GUI app): https://compbio.nyumc.org/icellr/
If you are using FlowJo or SeqGeq, they have made plugins for iCellR and other single cell tools: https://www.flowjo.com/exchange/#/ (list of all plugins) and https://www.flowjo.com/exchange/#/plugin/profile?id=34 (iCellR plugin). SeqGeq DE tutorial
For citing iCellR use this PMID: 34353854
iCellR publications: PMID: 34911733 (i.score and cell ranking) PMID 31744829 (scRNA-seq), PMID: 31934613 (bulk RNA-seq from TCGA), PMID: 32550269 (scVDJ-seq), PMID: 34135081, PMID: 33593073






# Install from CRAN
install.packages("iCellR")
# Install from github
#library(devtools)
#install_github("rezakj/iCellR")
# or
#git clone https://github.com/rezakj/iCellR.git
#R
#install.packages('iCellR/', repos = NULL, type="source")- Download and unzip a publicly available sample PBMC scRNA-Seq data.
# set your working directory
setwd("/your/download/directory")
# save the URL as an object
sample.file.url = "https://genome.med.nyu.edu/results/external/iCellR/data/pbmc3k_filtered_gene_bc_matrices.tar.gz"
# download the file
download.file(url = sample.file.url,
destfile = "pbmc3k_filtered_gene_bc_matrices.tar.gz",
method = "auto")
# unzip the file.
untar("pbmc3k_filtered_gene_bc_matrices.tar.gz")more data available here: https://genome.med.nyu.edu/results/external/iCellR/

To run a test sample follow these steps:
- Go to the R environment load the iCellR package and the PBMC sample data that you downloaded.
library("iCellR")
my.data <- load10x("filtered_gene_bc_matrices/hg19/")
# This directory includes; barcodes.tsv, genes.tsv/features.tsv and matrix.mtx files
# Data could be zipped or unzipped.
# if your data is in a csv or tsv format read it like this example
# my.data <- read.delim("CITE-Seq_sample_RNA.tsv.gz",header=TRUE)
# if your data is in a h5 format read it like this example
# data <- load.h5("filtered_feature_bc_matrix.h5")To see the help page for each function use question mark as:
?load10x- Aggregate data
Conditions in iCellR are set or shown in the column names of the data and are separated by an underscore "_" sign. Let's say you want to merge multiple datasets (data frames/matrices) into one file and run iCellR in aggregate mode (all samples together). You can do so using "data.aggregation" function. Here’s an example: I divided this sample into four datasets and then aggregated them into one matrix. Here we are assuming you have four samples (e.g. WT,KO,Ctrl,KD). In this way, iCellR will know you have 4 samples for the rest of the analysis (e.g. batch alignment, plots, DE, etc.).
dim(my.data)
# [1] 32738 2700
# divide your sample into three samples for this example
sample1 <- my.data[1:900]
sample2 <- my.data[901:1800]
sample3 <- my.data[1801:2300]
sample4 <- my.data[2301:2700]
# merge all of your samples to make a single aggregated file.
my.data <- data.aggregation(samples = c("sample1","sample2","sample3","sample4"),
condition.names = c("WT","KO","Ctrl","KD"))- Check the head of your file.
# here is how the head of the first 2 cells in the aggregated file looks like.
head(my.data)[1:2]
# WT_AAACATACAACCAC-1 WT_AAACATTGAGCTAC-1
#A1BG 0 0
#A1BG.AS1 0 0
#A1CF 0 0
#A2M 0 0
#A2M.AS1 0 0
# as you see the header has the conditions now- Make an object of class iCellR.
my.obj <- make.obj(my.data)
my.obj
###################################
,--. ,-----. ,--.,--.,------.
`--'' .--./ ,---. | || || .--. '
,--.| | | .-. :| || || '--'.'
| |' '--'\ --. | || || |
`--' `-----' `----'`--'`--'`--' '--'
###################################
An object of class iCellR version: 1.6.0
Raw/original data dimentions (rows,columns): 32738,2700
Data conditions in raw data: Ctrl,KD,KO,WT (500,400,900,900)
Row names: A1BG,A1BG.AS1,A1CF ...
Columns names: WT_AAACATACAACCAC.1,WT_AAACATTGAGCTAC.1,WT_AAACATTGATCAGC.1 ...
###################################
QC stats performed:FALSE, PCA performed:FALSE
Clustering performed:FALSE, Number of clusters:0
tSNE performed:FALSE, UMAP performed:FALSE, DiffMap performed:FALSE
Main data dimensions (rows,columns): 0,0
Normalization factors:,...
Imputed data dimensions (rows,columns):0,0
############## scVDJ-seq ###########
VDJ data dimentions (rows,columns):0,0
############## CITE-seq ############
ADT raw data dimensions (rows,columns):0,0
ADT main data dimensions (rows,columns):0,0
ADT columns names:...
ADT row names:...
############## scATAC-seq ############
ATAC raw data dimensions (rows,columns):0,0
ATAC main data dimensions (rows,columns):0,0
ATAC columns names:...
ATAC row names:...
############## Spatial ###########
Spatial data dimentions (rows,columns):0,0
########### iCellR object ##########- Perform some QC
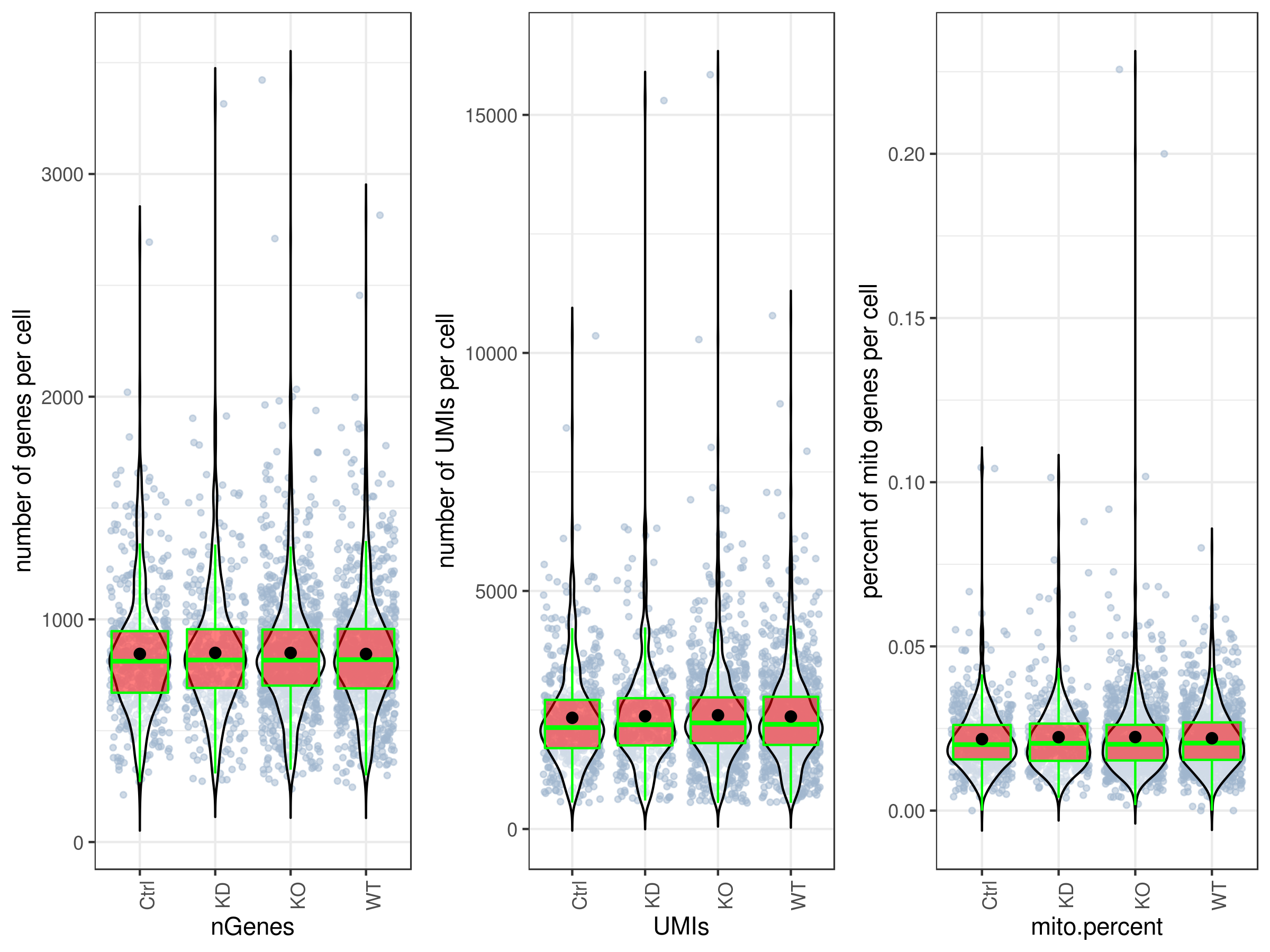
my.obj <- qc.stats(my.obj)- Plot QC
By default all the plotting functions would create interactive html files unless you set this parameter: interactive = FALSE.
# plot UMIs, genes and percent mito all at once and in one plot.
# you can make them individually as well, see the arguments ?stats.plot.
stats.plot(my.obj,
plot.type = "three.in.one",
out.name = "UMI-plot",
interactive = FALSE,
cell.color = "slategray3",
cell.size = 1,
cell.transparency = 0.5,
box.color = "red",
box.line.col = "green")
# Scatter plots
stats.plot(my.obj, plot.type = "point.mito.umi", out.name = "mito-umi-plot")
stats.plot(my.obj, plot.type = "point.gene.umi", out.name = "gene-umi-plot")


- Filter cells.
iCellR allows you to filter based on library sizes (UMIs), number of genes per cell, percent mitochondrial content, one or more genes, and cell ids.
my.obj <- cell.filter(my.obj,
min.mito = 0,
max.mito = 0.05,
min.genes = 200,
max.genes = 2400,
min.umis = 0,
max.umis = Inf)
#[1] "cells with min mito ratio of 0 and max mito ratio of 0.05 were filtered."
#[1] "cells with min genes of 200 and max genes of 2400 were filtered."
#[1] "No UMI number filter"
#[1] "No cell filter by provided gene/genes"
#[1] "No cell id filter"
#[1] "filters_set.txt file has beed generated and includes the filters set for this experiment."
# more examples
# my.obj <- cell.filter(my.obj, filter.by.gene = c("RPL13","RPL10")) # filter our cell having no counts for these genes
# my.obj <- cell.filter(my.obj, filter.by.cell.id = c("WT_AAACATACAACCAC.1")) # filter our cell cell by their cell ids.
# chack to see how many cells are left.
dim(my.obj@main.data)
#[1] 32738 2637- Down sampling
This step is optional and is for having the same number of cells for each condition.
# optional
# my.obj <- down.sample(my.obj)
#[1] "From"
#[1] "Data conditions: Ctrl,KO,WT (877,877,883)"
#[1] "to"
#[1] "Data conditions: Ctrl,KO,WT (877,877,877)"- Normalize data
You have a few options to normalize your data based on your study. You can also normalize your data using tools other than iCellR and import your data to iCellR. We recommend "ranked.glsf" normalization for most single cell studies. This normalization is great for fixing matrixes with lots of zeros and because it's geometric it will reduce some of batch differences in the library sizes, as long as all the data is aggregated into one file (to aggregate your data see "aggregating data" section above). GLSF stands for Geometric Library Size Factor, this is very similar to the normalization done by DESeq2 and the ranked part would take the sum of the top most expressed genes as your library size instead of the full LB size which is to help resduce some of the drop out effects on normalization.
my.obj <- norm.data(my.obj,
norm.method = "ranked.glsf",
top.rank = 500) # best for scRNA-Seq
# more examples
#my.obj <- norm.data(my.obj, norm.method = "ranked.deseq", top.rank = 500)
#my.obj <- norm.data(my.obj, norm.method = "deseq") # best for bulk RNA-Seq
#my.obj <- norm.data(my.obj, norm.method = "global.glsf") # best for bulk RNA-Seq
#my.obj <- norm.data(my.obj, norm.method = "rpm", rpm.factor = 100000) # best for bulk RNA-Seq
#my.obj <- norm.data(my.obj, norm.method = "spike.in", spike.in.factors = NULL)
#my.obj <- norm.data(my.obj, norm.method = "no.norm") # if the data is already normalized- Perform second QC (optioal)
#my.obj <- qc.stats(my.obj,which.data = "main.data")
#stats.plot(my.obj,
# plot.type = "all.in.one",
# out.name = "UMI-plot",
# interactive = F,
# cell.color = "slategray3",
# cell.size = 1,
# cell.transparency = 0.5,
# box.color = "red",
# box.line.col = "green",
# back.col = "white")- Scale data (optional)
iCellR does not need this step as it scales the data when they need to be scaled on the fly; like for plotting or running PCA. This is because, it is important to use the untransformed data for differential expression analysis to calculate the accurate/true fold changes. If you run this function the scaled data will replace the main data for this reason and instead will be saved in different data slot in the object for you to download if you need it for plotting or other reasons.
# my.obj <- data.scale(my.obj)- Gene stats
my.obj <- gene.stats(my.obj, which.data = "main.data")
head(my.obj@gene.data[order(my.obj@gene.data$numberOfCells, decreasing = T),])
# genes numberOfCells totalNumberOfCells percentOfCells meanExp
#30303 TMSB4X 2637 2637 100.00000 38.55948
#3633 B2M 2636 2637 99.96208 45.07327
#14403 MALAT1 2636 2637 99.96208 70.95452
#27191 RPL13A 2635 2637 99.92416 32.29009
#27185 RPL10 2632 2637 99.81039 35.43002
#27190 RPL13 2630 2637 99.73455 32.32106
# SDs condition
#30303 7.545968e-15 all
#3633 2.893940e+01 all
#14403 7.996407e+01 all
#27191 2.783799e+01 all
#27185 2.599067e+01 all
#27190 2.661361e+01 all- Make a gene model for clustering
This function will help you find a good number of genes to use for running PCA.
# See model plot
make.gene.model(my.obj, my.out.put = "plot",
dispersion.limit = 1.5,
base.mean.rank = 500,
no.mito.model = T,
mark.mito = T,
interactive = F,
out.name = "gene.model")
# Write the gene model data into the object
my.obj <- make.gene.model(my.obj, my.out.put = "data",
dispersion.limit = 1.5,
base.mean.rank = 500,
no.mito.model = T,
mark.mito = T,
interactive = F,
out.name = "gene.model")
head(my.obj@gene.model)
# "ACTB" "ACTG1" "ACTR3" "AES" "AIF1" "ALDOA"
# get html plot (optional)
#make.gene.model(my.obj, my.out.put = "plot",
# dispersion.limit = 1.5,
# base.mean.rank = 500,
# no.mito.model = T,
# mark.mito = T,
# interactive = T,
# out.name = "plot4_gene.model")To view an the html interactive plot click on this links: Dispersion plot

- Perform Principal component analysis (PCA)
Note: skip this step if you plan to do batch correction. For batch correction (sample alignment/harmonization/integration) see the sections; CPCA, CCCA, MNN or anchor alignment.
# If you run PCA (run.pca) there would be no batch alignment but if you run CPCA (using iba function) this would perform batch alignment and PCA after batch alignment. Example for batch alignment using iba function:
# my.obj <- iba(my.obj,dims = 1:30, k = 10,ba.method = "CPCA", method = "gene.model", gene.list = [email protected])
# run PCA in case no batch alignment is necessary
my.obj <- run.pca(my.obj, method = "gene.model", gene.list = my.obj@gene.model,data.type = "main")
opt.pcs.plot(my.obj)
# 2 round PCA (optional)
# This is to find top genes in the first 10 PCs and re-run PCA for better clustering.
## This is optional and might not be good in some cases
#length([email protected])
# 683
#my.obj <- find.dim.genes(my.obj, dims = 1:10,top.pos = 20, top.neg = 20) # (optional)
#length([email protected])
# 211
# second round PC
#my.obj <- run.pca(my.obj, method = "gene.model", gene.list = [email protected],data.type = "main")

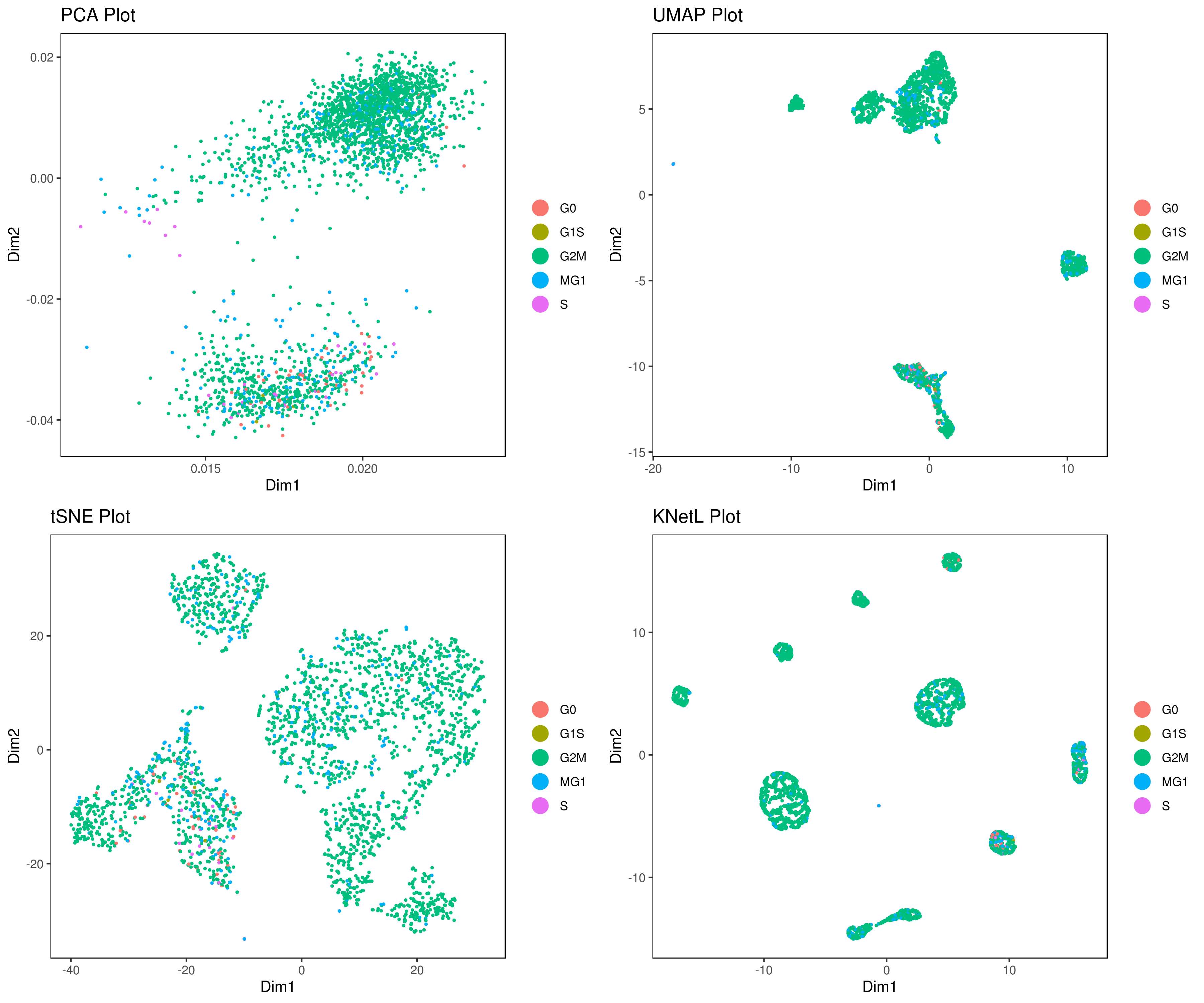
- Perform other dimensionality reductions (tSNE, UMAP, KNetL, PHATE, destiny, diffusion maps)
We recommend tSNE, UMAP and KNetL. KNetL is fundamentally more powerful.
# tSNE
my.obj <- run.pc.tsne(my.obj, dims = 1:10)
# UMAP
my.obj <- run.umap(my.obj, dims = 1:10)
# KNetL (for lager than 5000 cell use a zoom of about 400)
# Because knetl has a very high resolution it's best to use a dim of 20 (this usually works best for most data)
my.obj <- run.knetl(my.obj, dims = 1:20, zoom = 110, dim.redux = "umap") # (Important note!) don't forget to set the zoom in the right range
########################## IMPORTANT DISCLAIMER NOTE ###########################
*** KNetL map is very dynamic with zoom and dims! ***
*** Therefore it needs to be adjusted! ***
# For data with less than 1000 cells use a zoom of about 5-50.
# For data with 1000-5000 cells use a zoom of about 50-200.
# For data with 5000-10000 cells use a zoom of about 100-300.
# For data with 10000-30000 cells use a zoom of about 200-500.
# For data with more than 30000 cells use a zoom of about 400-600.
# zoom 400 is usually good for big data but adjust for intended resolution.
# Lower number for zoom in and higher for zoom out (its reverse).
# dims = 1:20 is generally good for most data.
# other parameters are best as default.
#### Just like a microscope, you need to zoom to see the intended amount of details.
#### Here we use a zoom of 100 or 110 but this might not be ideal for your data.
#### example: # my.obj <- run.knetl(my.obj, dims = 1:20, zoom = 400)
#### Because knetl has a very high resolution it's best to use a dim of 20 (this usually works best for most data)
###################################################################################
###################################################################################
###################################################################################
###################################################################################
# diffusion map
# this requires python packge phate or bioconductor R package destiny
# How to install destiny
# if (!requireNamespace("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
# BiocManager::install("destiny")
# How to install phate
# pip install --user phate
# Install phateR version 2.9
# wget https://cran.r-project.org/src/contrib/Archive/phateR/phateR_0.2.9.tar.gz
# install.packages('phateR/', repos = NULL, type="source")
# or
# library(devtools)
# install_version("phateR", version = "0.2.9", repos = "http://cran.us.r-project.org")
# optional
# library(destiny)
# my.obj <- run.diffusion.map(my.obj, dims = 1:10)
# or
# library(phateR)
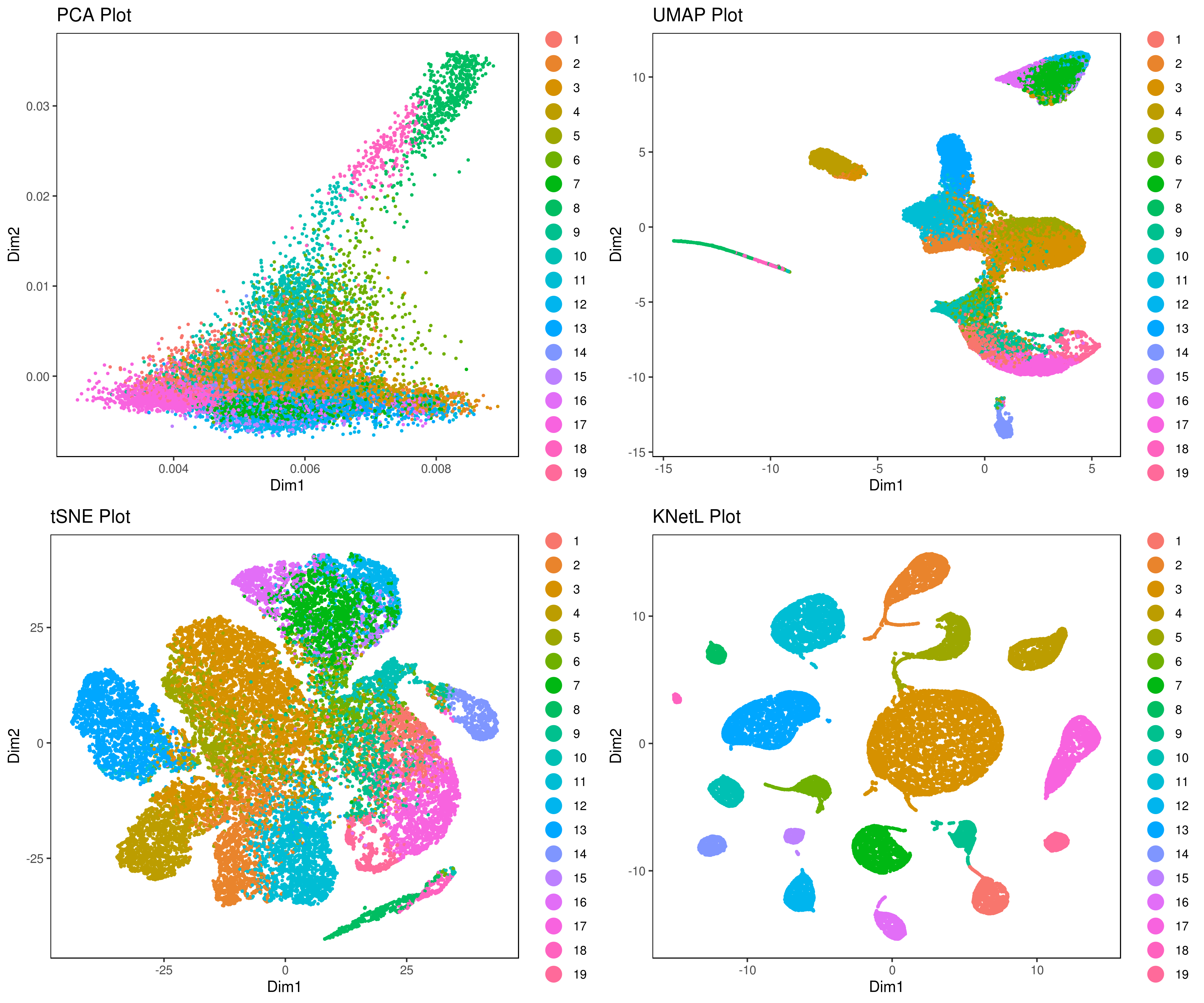
# my.obj <- run.diffusion.map(my.obj, dims = 1:10, method = "phate")- Visualizing the results of dimensionality reductions before clustering (optional)
A= cluster.plot(my.obj,plot.type = "pca",interactive = F)
B= cluster.plot(my.obj,plot.type = "umap",interactive = F)
C= cluster.plot(my.obj,plot.type = "tsne",interactive = F)
D= cluster.plot(my.obj,plot.type = "knetl",interactive = F)
library(gridExtra)
grid.arrange(A,B,C,D)
We provide three functions to run the clustering method of your choice:
Faster and optimized for iCellR. This function takes PCA, UMAP or tSNE, Destiny (diffusion map), PHATE or KNetL map as input. This function is using Louvain algorithm for clustering a graph made using KNN. Similar to PhenoGraph (Levine et al., Cell, 2015) however instead of Jaccard similarity values we use distance (euclidean by default) values for the weights.
R implementation of the PhenoGraph algorithm. Rphenograph wrapper (Levine et al., Cell, 2015).
In this function we provide a variety of many other options for you to explore the data with different flavours of clustering and indexing methods. Choose any combinations from the table below.
| clustering methods | distance methods | indexing methods |
|---|---|---|
| ward.D, ward.D2, single, complete, average, mcquitty, median, centroid, kmeans | euclidean, maximum, manhattan, canberra, binary, minkowski or NULL | kl, ch, hartigan, ccc, scott, marriot, trcovw, tracew, friedman, rubin, cindex, db, silhouette, duda, pseudot2, beale, ratkowsky, ball, ptbiserial, gap, frey, mcclain, gamma, gplus, tau, dunn, hubert, sdindex, dindex, sdbw |
Conventionally people cluster based on PCA data (usually first 10 dimensions) however you have the option of choosing tSNE, UMAP and KNetL map dimensions as well. If you have adjusted your KNetL map and are confident about the results we recommend clustering based on KNetL map.
This is one of the harder parts of the analysis and sometimes you need to adjust your clustering based on marker genes. This means you might need to merge some clusters, gate (see our cell gating tools) or try different sensitivities to find more or less communities.
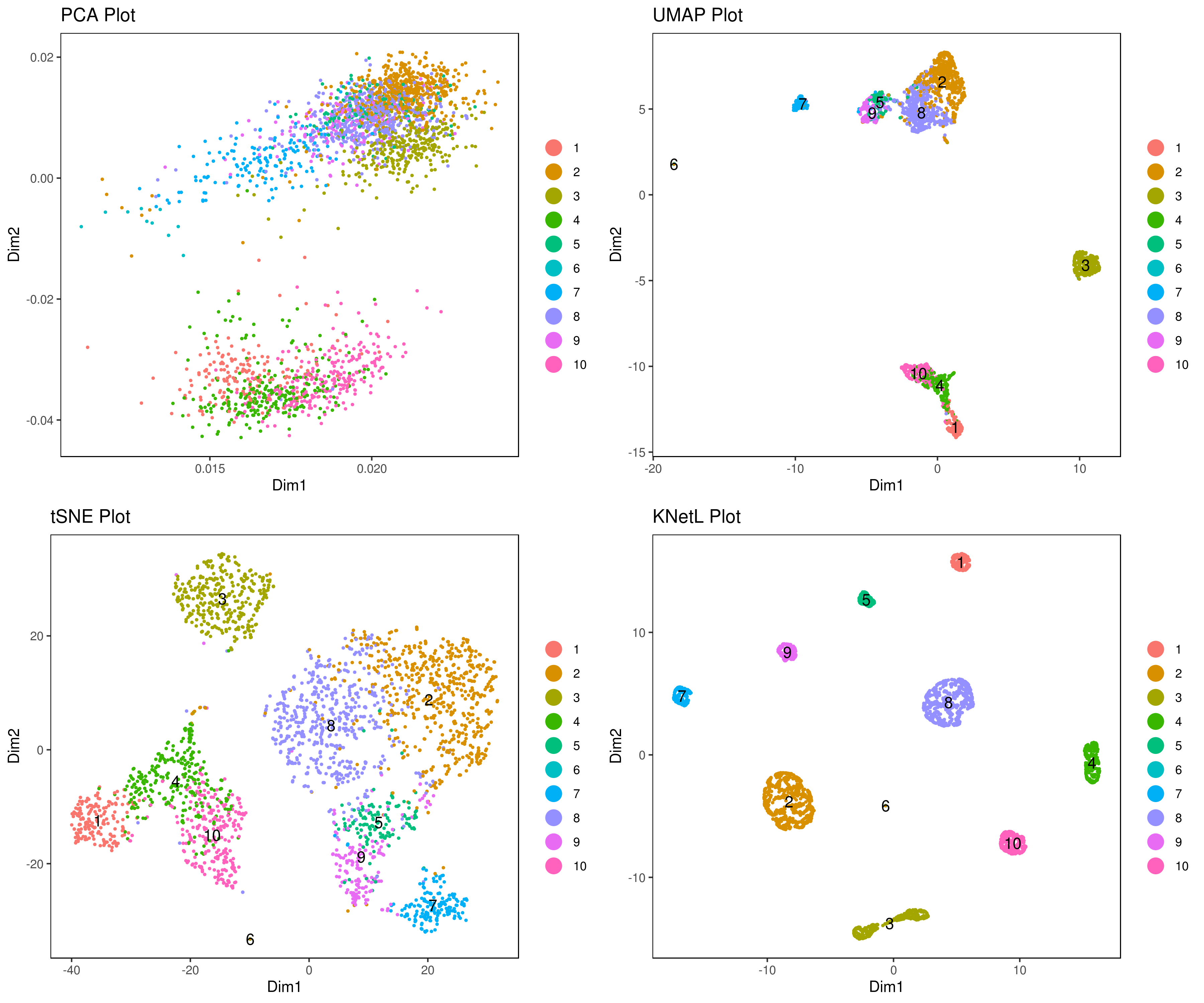
# clustering based on KNetL
my.obj <- iclust(my.obj, sensitivity = 150, data.type = "knetl")
# clustering based on PCA
# my.obj <- iclust(my.obj, sensitivity = 150, data.type = "pca", dims=1:10)
# play with k to get the clusters right. Usually 150 is good.
###### more examples
# clustering based on PCA
# my.obj <- iclust(my.obj,
# dist.method = "euclidean",
# sensitivity = 100,
# dims = 1:10,
# data.type = "pca")
# or
# run.phenograph
# my.obj <- run.phenograph(my.obj,k = 100,dims = 1:10)
# or
# run.clustering
# my.obj <- run.clustering(my.obj,
# clust.method = "kmeans",
# dist.method = "euclidean",
# index.method = "silhouette",
# max.clust = 25,
# min.clust = 2,
# dims = 1:10)
# If you want to manually set the number of clusters, and not used the predicted optimal number, set the minimum and maximum to the number you want:
#my.obj <- run.clustering(my.obj,
# clust.method = "ward.D",
# dist.method = "euclidean",
# index.method = "ccc",
# max.clust = 8,
# min.clust = 8,
# dims = 1:10)
# more examples
#my.obj <- run.clustering(my.obj,
# clust.method = "ward.D",
# dist.method = "euclidean",
# index.method = "kl",
# max.clust = 25,
# min.clust = 2,
# dims = 1:10)- Visualize data clustering results
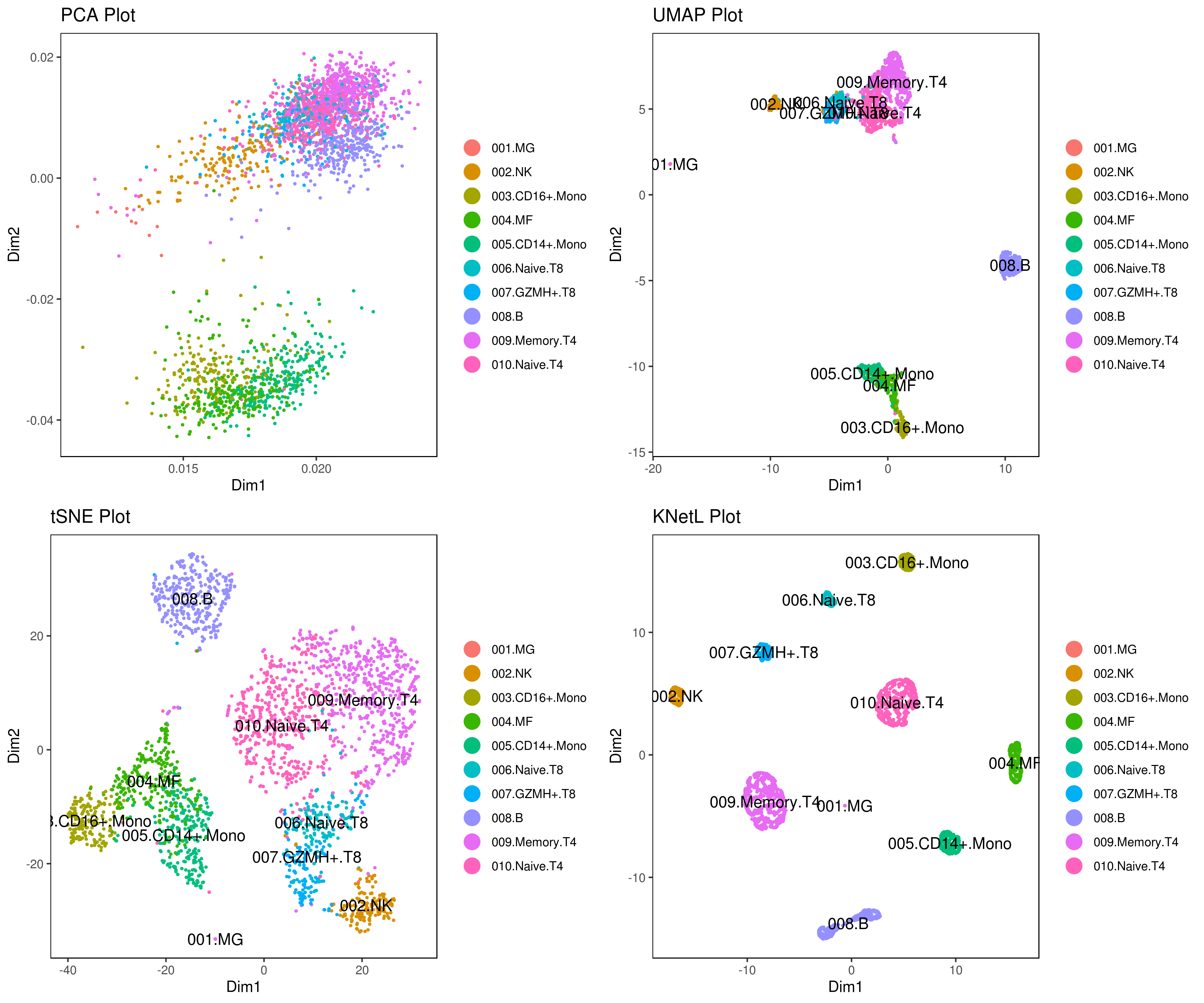
# plot clusters (in the figures below clustering is done based on KNetL)
# example: # my.obj <- iclust(my.obj, k = 150, data.type = "knetl")
A <- cluster.plot(my.obj,plot.type = "pca",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
B <- cluster.plot(my.obj,plot.type = "umap",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=T)
C <- cluster.plot(my.obj,plot.type = "tsne",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=T)
D <- cluster.plot(my.obj,plot.type = "knetl",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=T)
library(gridExtra)
grid.arrange(A,B,C,D)
- Re-numbering clusters based on their distances, this is so that the are more in consecutive order (optional)
This is visually helpful to look at your heatmap after finding marker genes and can help you decide which clusters need to be merged and adjusted.
my.obj <- clust.ord(my.obj,top.rank = 500, how.to.order = "distance")
#my.obj <- clust.ord(my.obj,top.rank = 500, how.to.order = "random")
A= cluster.plot(my.obj,plot.type = "pca",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
B= cluster.plot(my.obj,plot.type = "umap",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=T)
C= cluster.plot(my.obj,plot.type = "tsne",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=T)
D= cluster.plot(my.obj,plot.type = "knetl",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=T)
library(gridExtra)
grid.arrange(A,B,C,D)
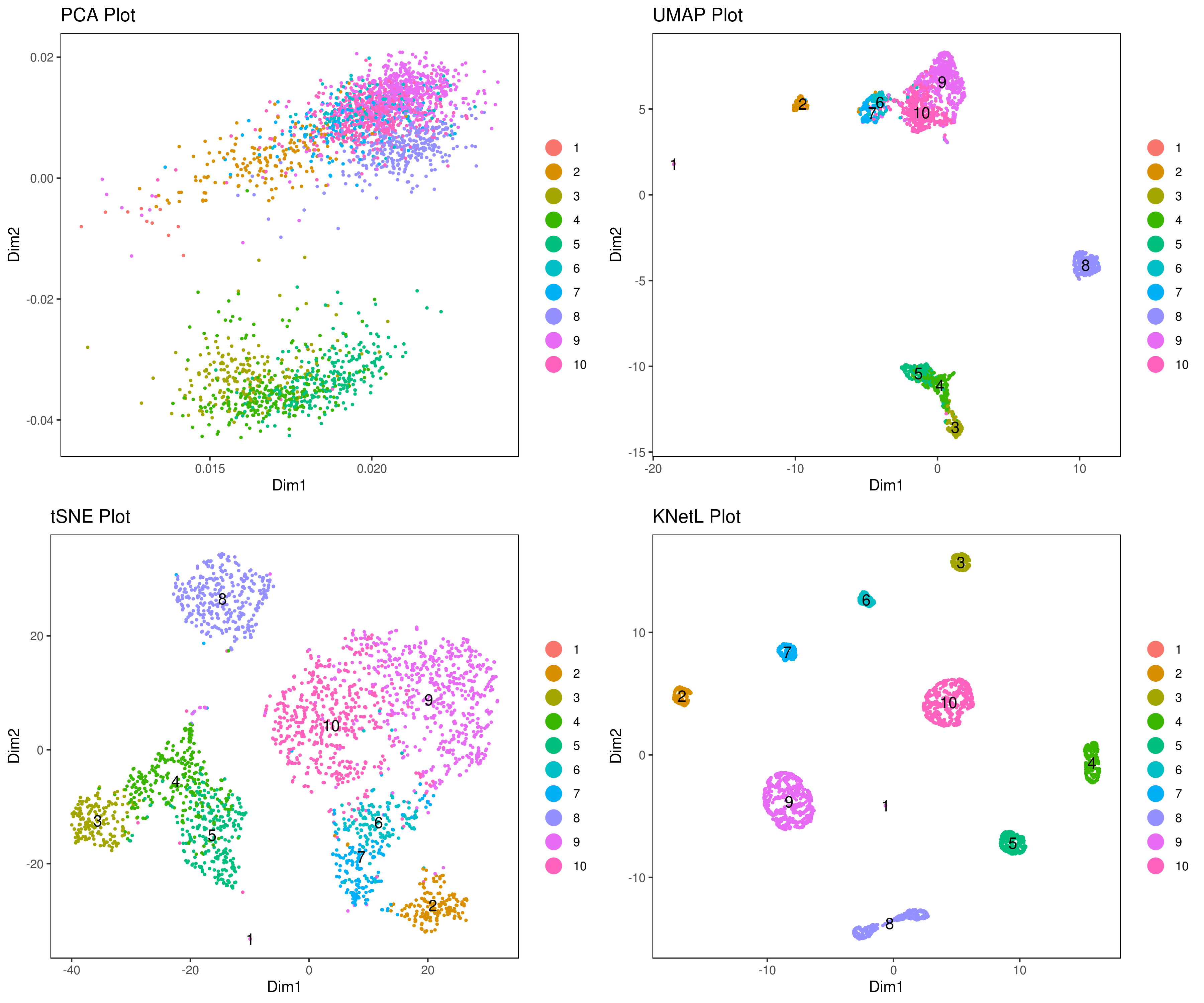
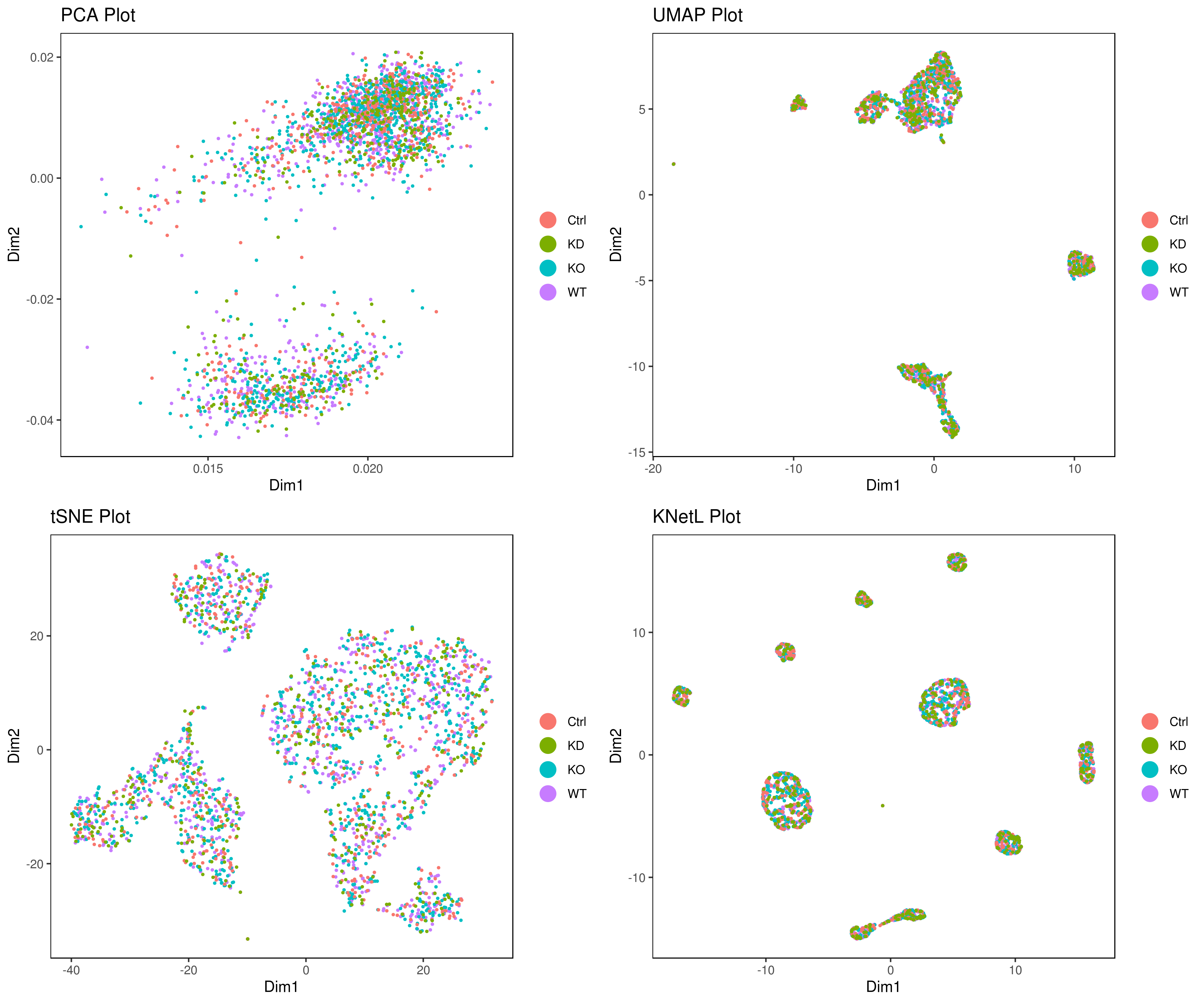
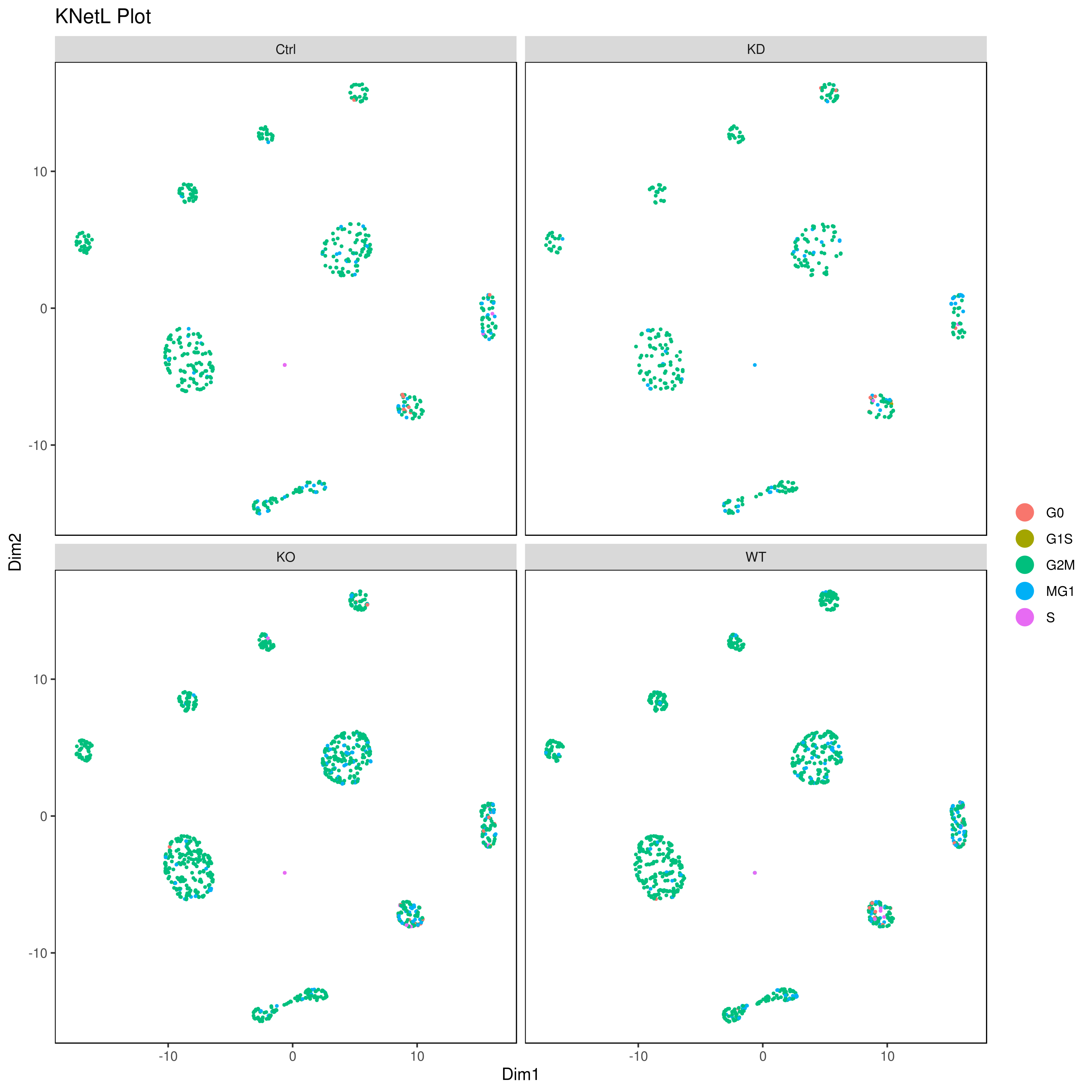
- Look at conditions
# conditions
A <- cluster.plot(my.obj,plot.type = "pca",col.by = "conditions",interactive = F,cell.size = 0.5)
B <- cluster.plot(my.obj,plot.type = "umap",col.by = "conditions",interactive = F,cell.size = 0.5)
C <- cluster.plot(my.obj,plot.type = "tsne",col.by = "conditions",interactive = F,cell.size = 0.5)
D <- cluster.plot(my.obj,plot.type = "knetl",col.by = "conditions",interactive = F,cell.size = 0.5)
library(gridExtra)
grid.arrange(A,B,C,D)
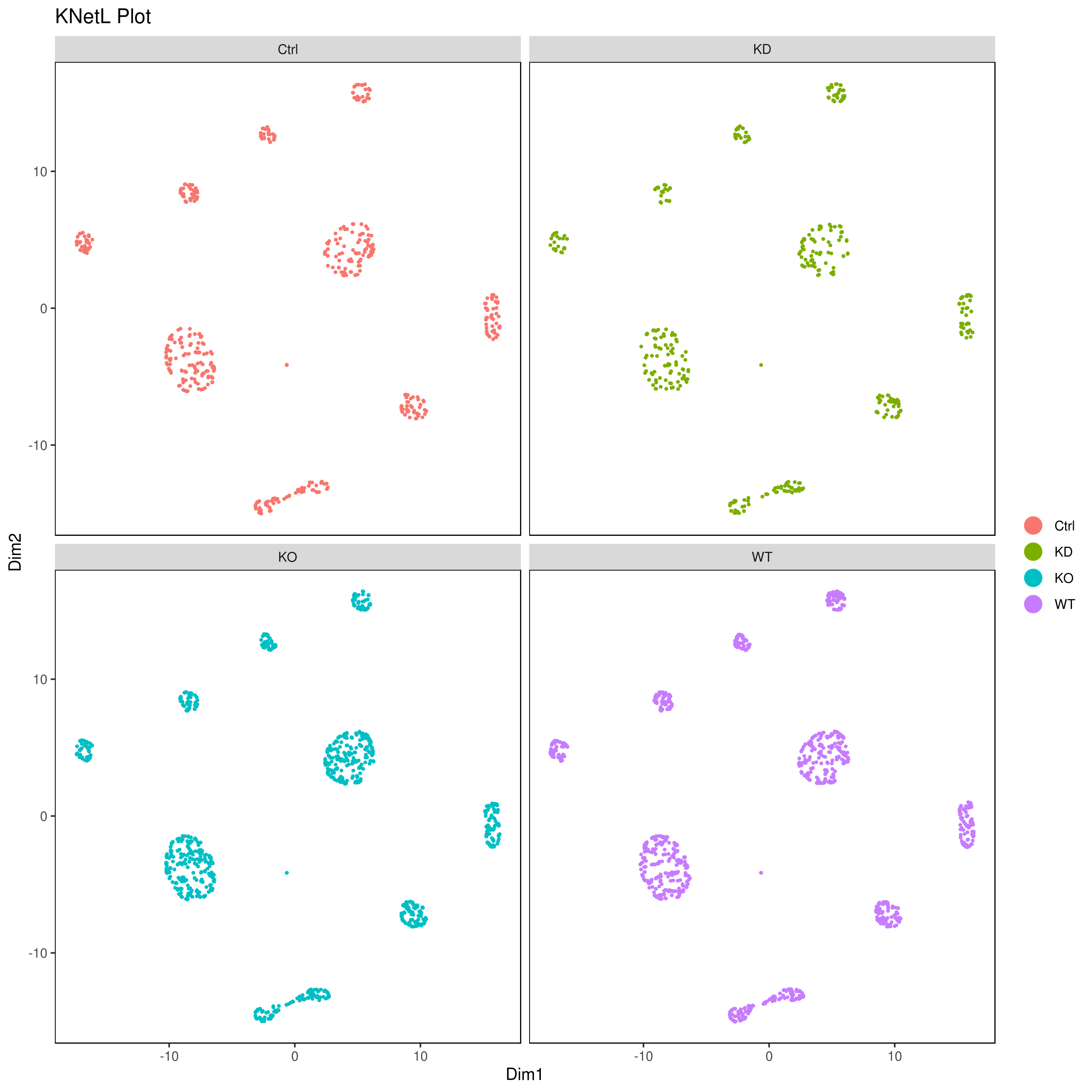
### or
cluster.plot(my.obj,
cell.size = 0.5,
plot.type = "knetl",
cell.color = "black",
back.col = "white",
col.by = "conditions",
cell.transparency = 1,
clust.dim = 2,
interactive = F,cond.facet = T)
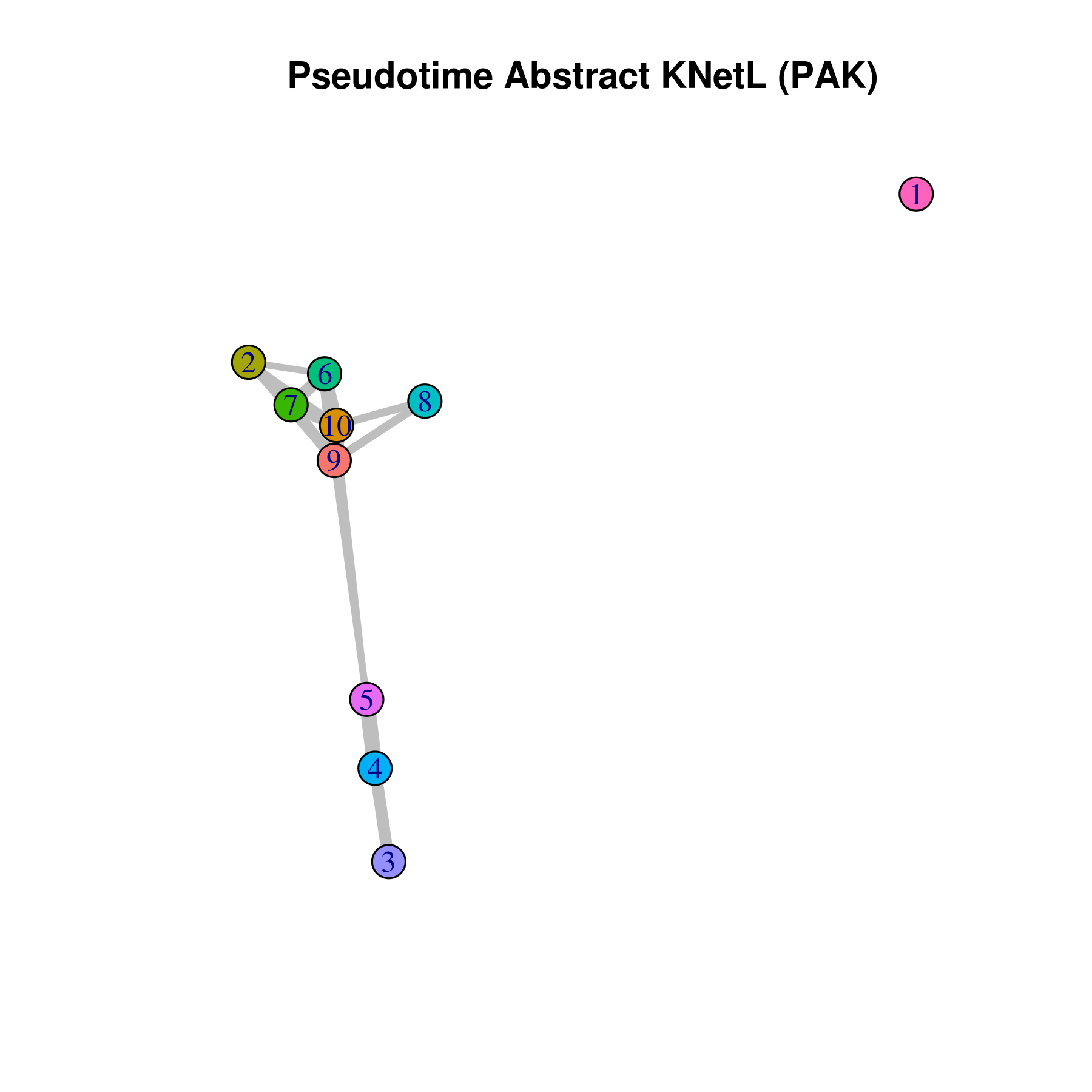
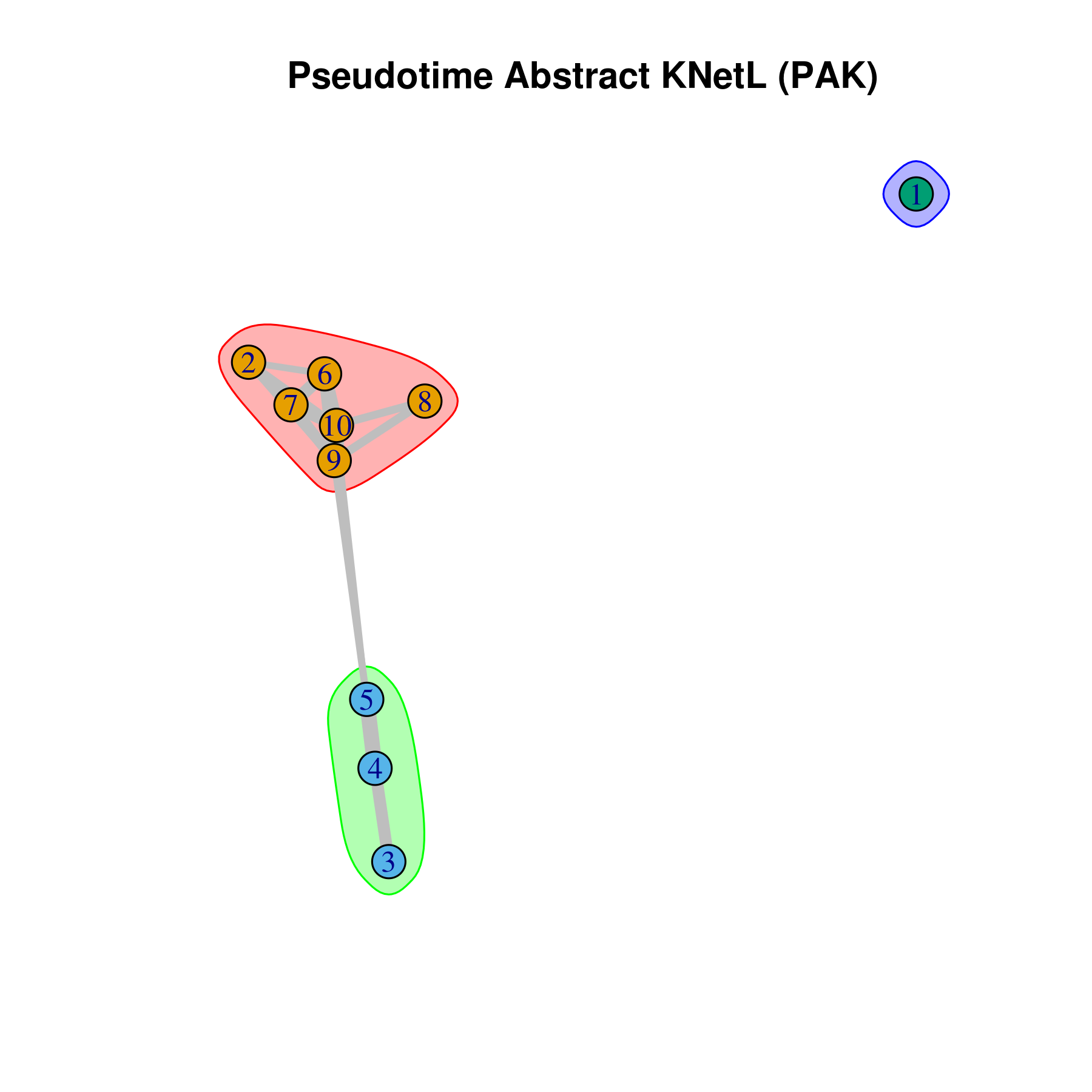
- Pseudotime Abstract KNetL map (PAK map)
This is very helpful to see the distances or similarities between different communities. The shorter and thicker the lines/links (rubber bands) are the more similar the communities. The nodes are the clusters and the edges or links are the distance between them.
pseudotime.knetl(my.obj,interactive = F,cluster.membership = F,conds.to.plot = NULL)
## with memberships
pseudotime.knetl(my.obj,interactive = F,cluster.membership = T,conds.to.plot = NULL)
### intractive plot
pseudotime.knetl(my.obj,interactive = T)
- Average expression per cluster
# for all cunditions
my.obj <- clust.avg.exp(my.obj, conds.to.avg = NULL)
# for one cundition
#my.obj <- clust.avg.exp(my.obj, conds.to.avg = "WT")
# for two cundition
#my.obj <- clust.avg.exp(my.obj, conds.to.avg = c("WT","KO"))
head(my.obj@clust.avg)
# gene cluster_1 cluster_2 cluster_3 cluster_4 cluster_5
#1 A1BG 0 0.034248447 0.029590643 0.076486590 0.090270833
#2 A1BG.AS1 0 0.000000000 0.006274854 0.019724138 0.004700000
#3 A1CF 0 0.000000000 0.000000000 0.000000000 0.000000000
#4 A2M 0 0.006925466 0.003614035 0.000000000 0.000000000
#5 A2M.AS1 0 0.056155280 0.000000000 0.005344828 0.006795833
#6 A2ML1 0 0.000000000 0.000000000 0.000000000 0.000000000
# cluster_6 cluster_7 cluster_8 cluster_9 cluster_10
#1 0.074360294 0.07623494 0.04522321 0.088735057 0.065292818
#2 0.000000000 0.00000000 0.01553869 0.013072698 0.013550645
#3 0.000000000 0.00000000 0.00000000 0.000000000 0.000000000
#4 0.000000000 0.00000000 0.00000000 0.001810985 0.003200737
#5 0.008191176 0.06227108 0.00000000 0.011621971 0.012837937
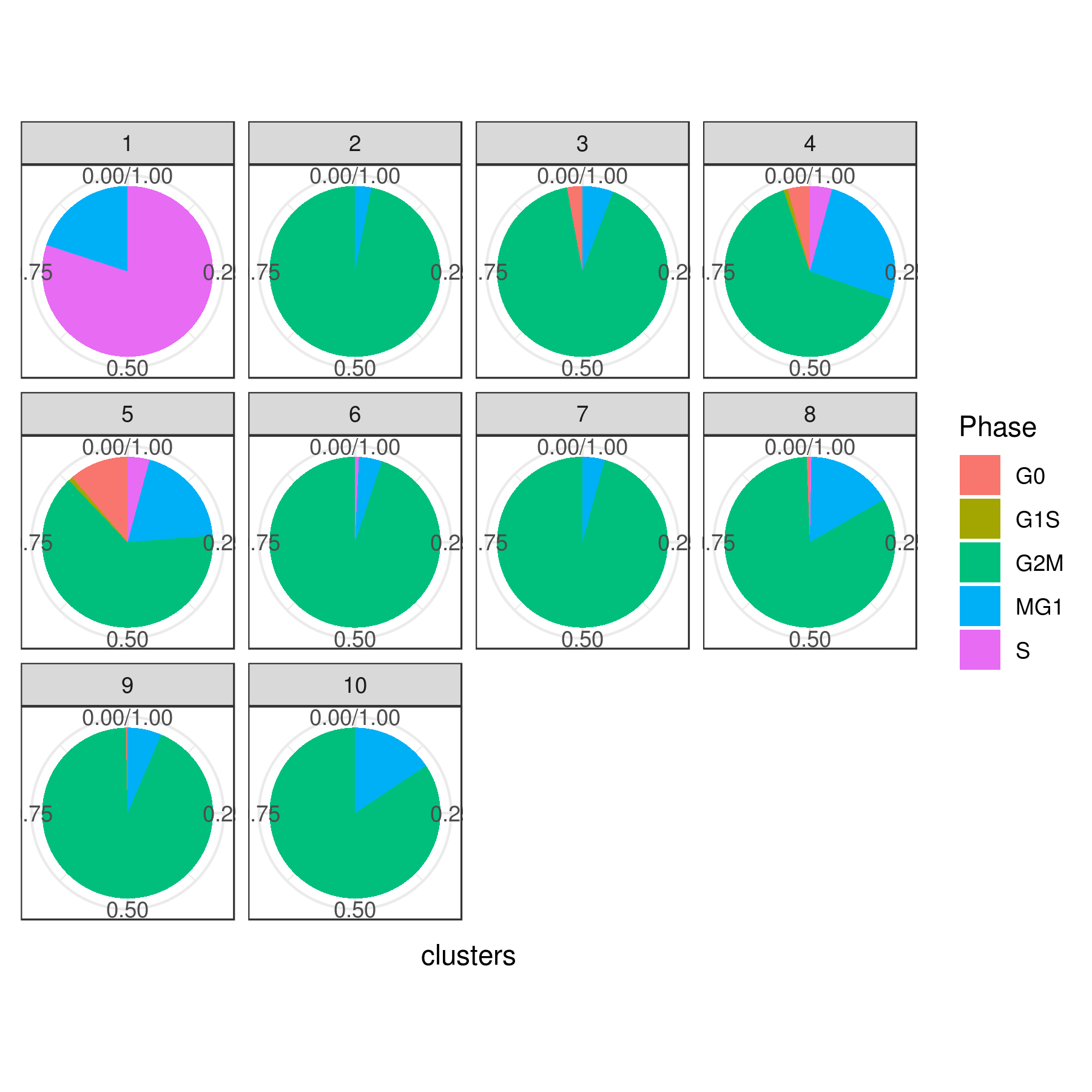
#6 0.000000000 0.00000000 0.00000000 0.000000000 0.000000000- Cell cycle prediction
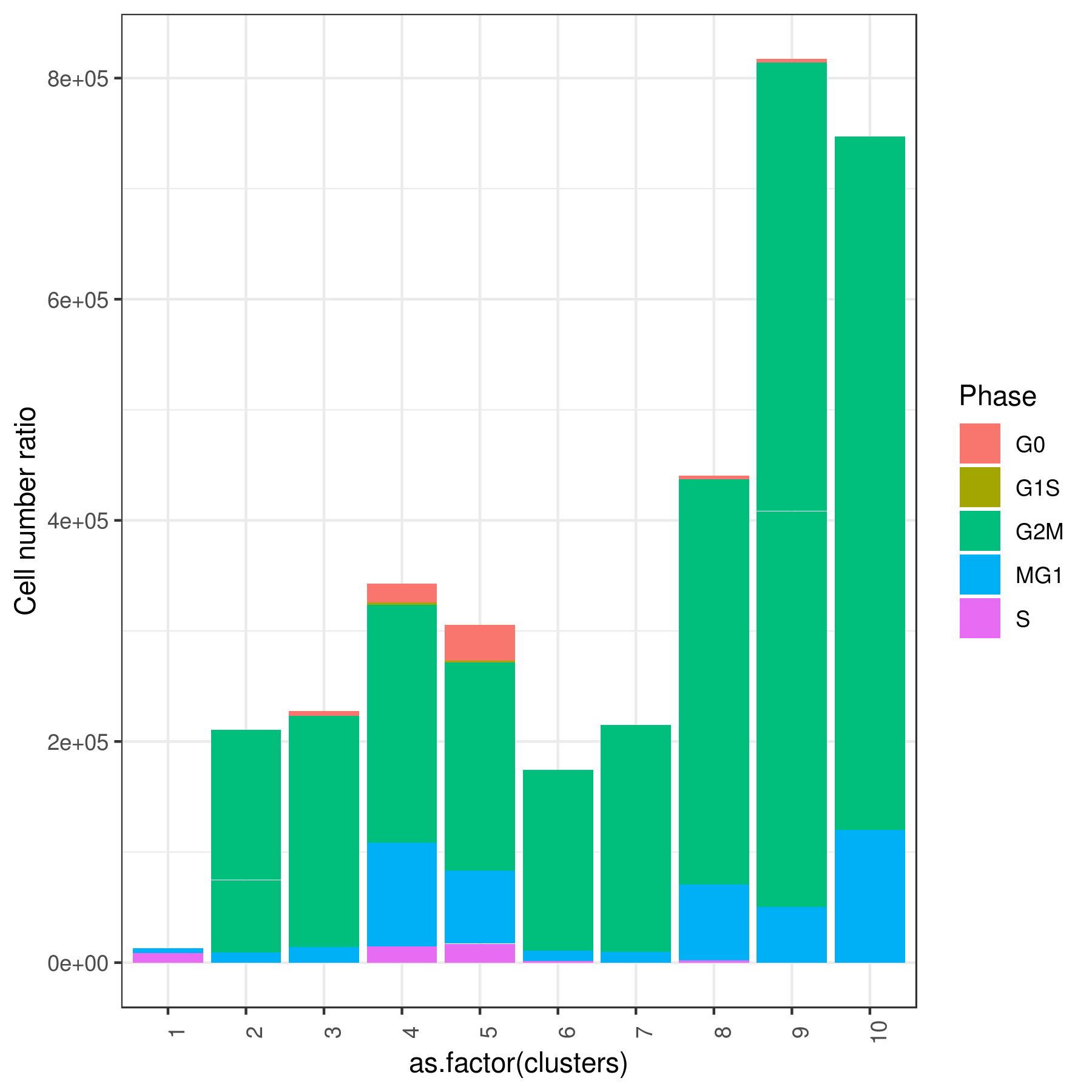
Tirosh scoring method Tirosh, et. al. 2016 (default) or coverage is used to calculate G0, G1S, G2M, M, G1M and S phase score. The gene lists for G0, G1S, G2M, M, G1M and S phase are chosen from previously published article Xue, et.al 2020
NOTE: These genes work best for cancer cells. You can use a different gene set for each category (G0, G1S, G2M, M, G1M and S).
# old method
# my.obj <- cc(my.obj, s.genes = s.phase, g2m.genes = g2m.phase)
# new method
G0 <- readLines(system.file('extdata', 'G0.txt', package = 'iCellR'))
G1S <- readLines(system.file('extdata', 'G1S.txt', package = 'iCellR'))
G2M <- readLines(system.file('extdata', 'G2M.txt', package = 'iCellR'))
M <- readLines(system.file('extdata', 'M.txt', package = 'iCellR'))
MG1 <- readLines(system.file('extdata', 'MG1.txt', package = 'iCellR'))
S <- readLines(system.file('extdata', 'S.txt', package = 'iCellR'))
# Tirosh scoring method (recomanded)
my.obj <- cell.cycle(my.obj, scoring.List = c("G0","G1S","G2M","M","MG1","S"), scoring.method = "tirosh")
# Coverage scoring method (recomanded)
# my.obj <- cell.cycle(my.obj, scoring.List = c("G0","G1S","G2M","M","MG1","S"), scoring.method = "coverage")
# plot cell cycle
A= cluster.plot(my.obj,plot.type = "pca",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T,col.by = "cc")
B= cluster.plot(my.obj,plot.type = "umap",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=T, col.by = "cc")
C= cluster.plot(my.obj,plot.type = "tsne",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=T, col.by = "cc")
D= cluster.plot(my.obj,plot.type = "knetl",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=T, col.by = "cc")
library(gridExtra)
grid.arrange(A,B,C,D)
## or
cluster.plot(my.obj,
cell.size = 0.5,
plot.type = "knetl",
col.by = "cc",
cell.color = "black",
back.col = "white",
cell.transparency = 1,
clust.dim = 2,
interactive = F,cond.facet = T)
# Pie
clust.stats.plot(my.obj, plot.type = "pie.cc", interactive = F, conds.to.plot = NULL)
dev.off()
# bar
clust.stats.plot(my.obj, plot.type = "bar.cc", interactive = F, conds.to.plot = NULL)
dev.off()
# or per condition
# clust.stats.plot(my.obj, plot.type = "pie.cc", interactive = F, conds.to.plot = "WT")
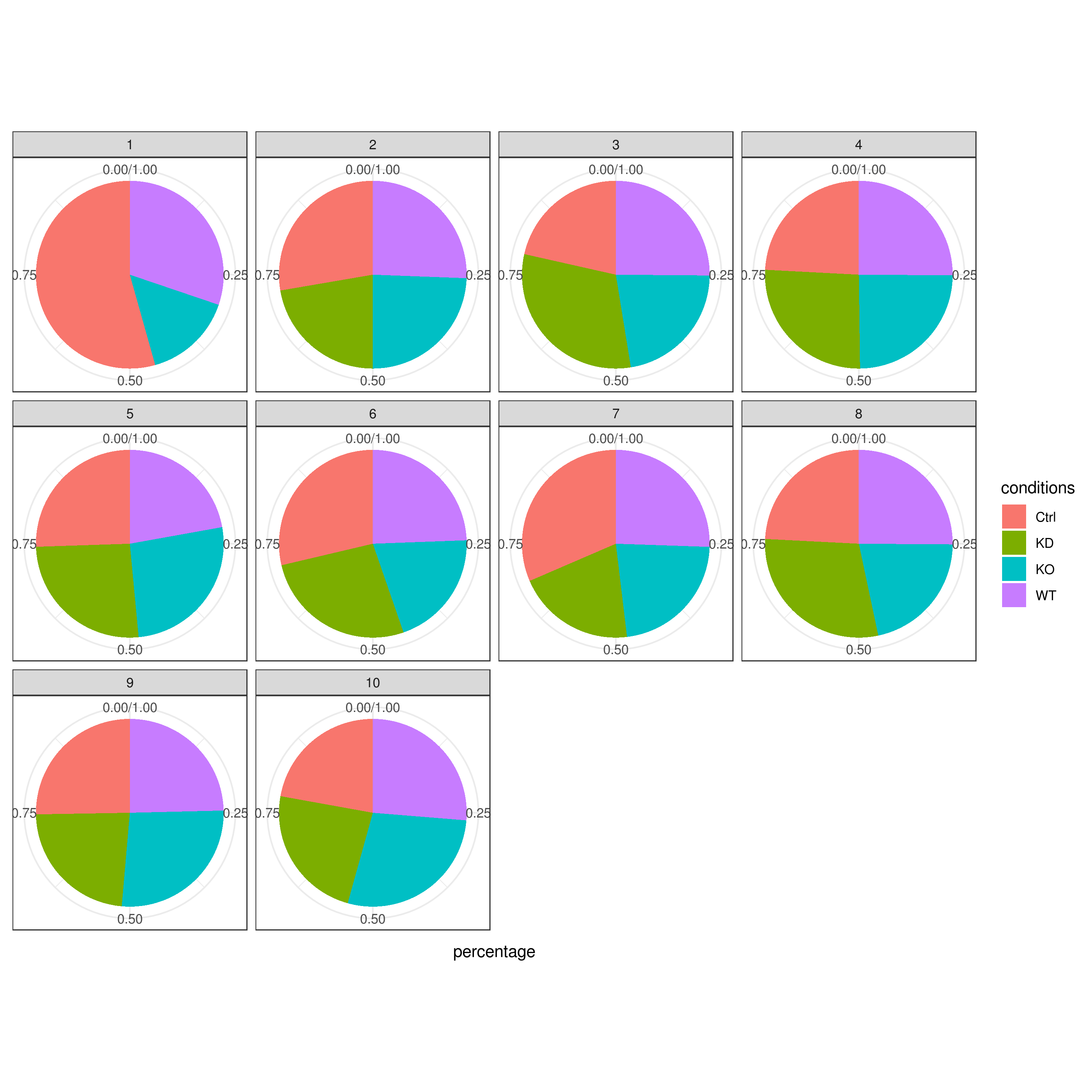
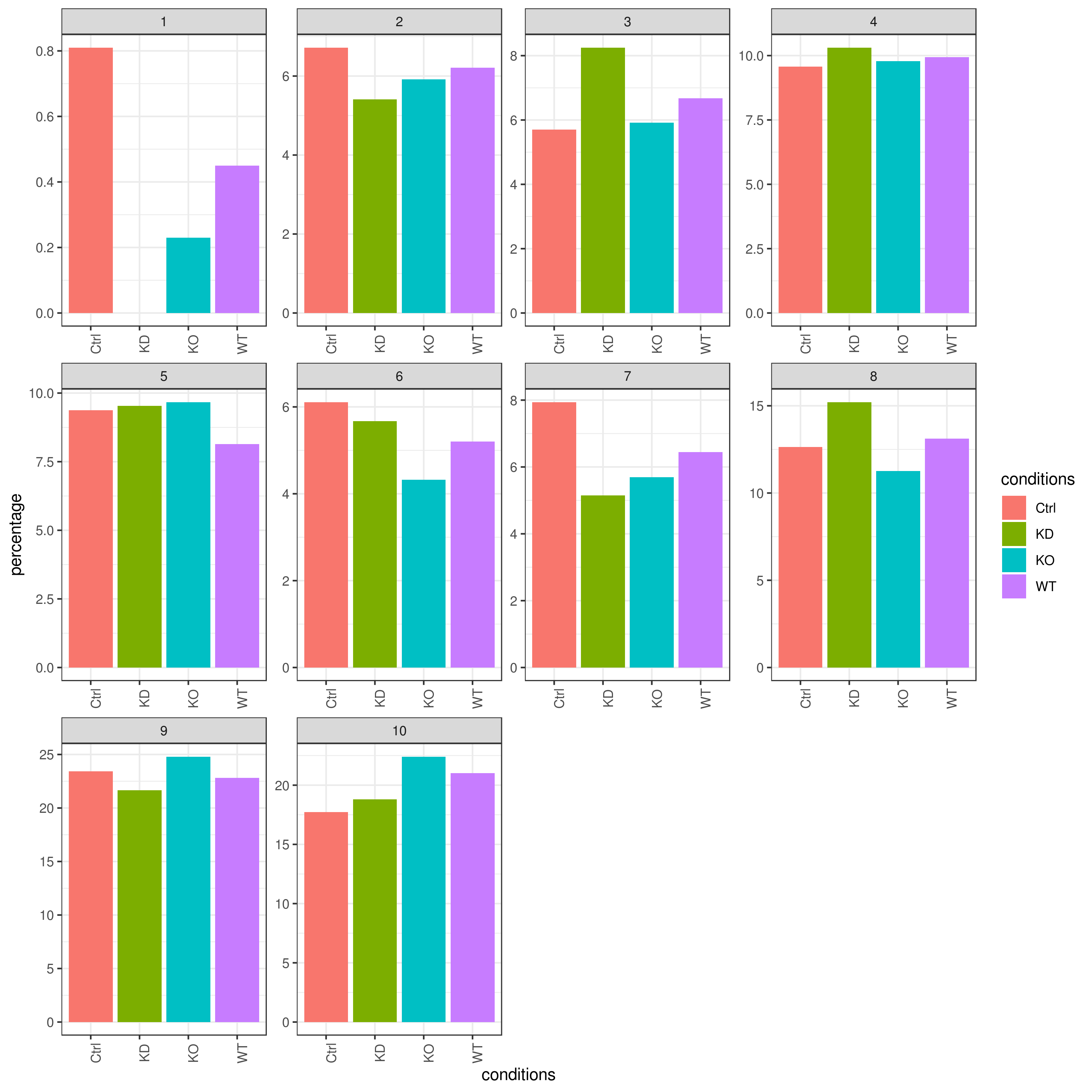
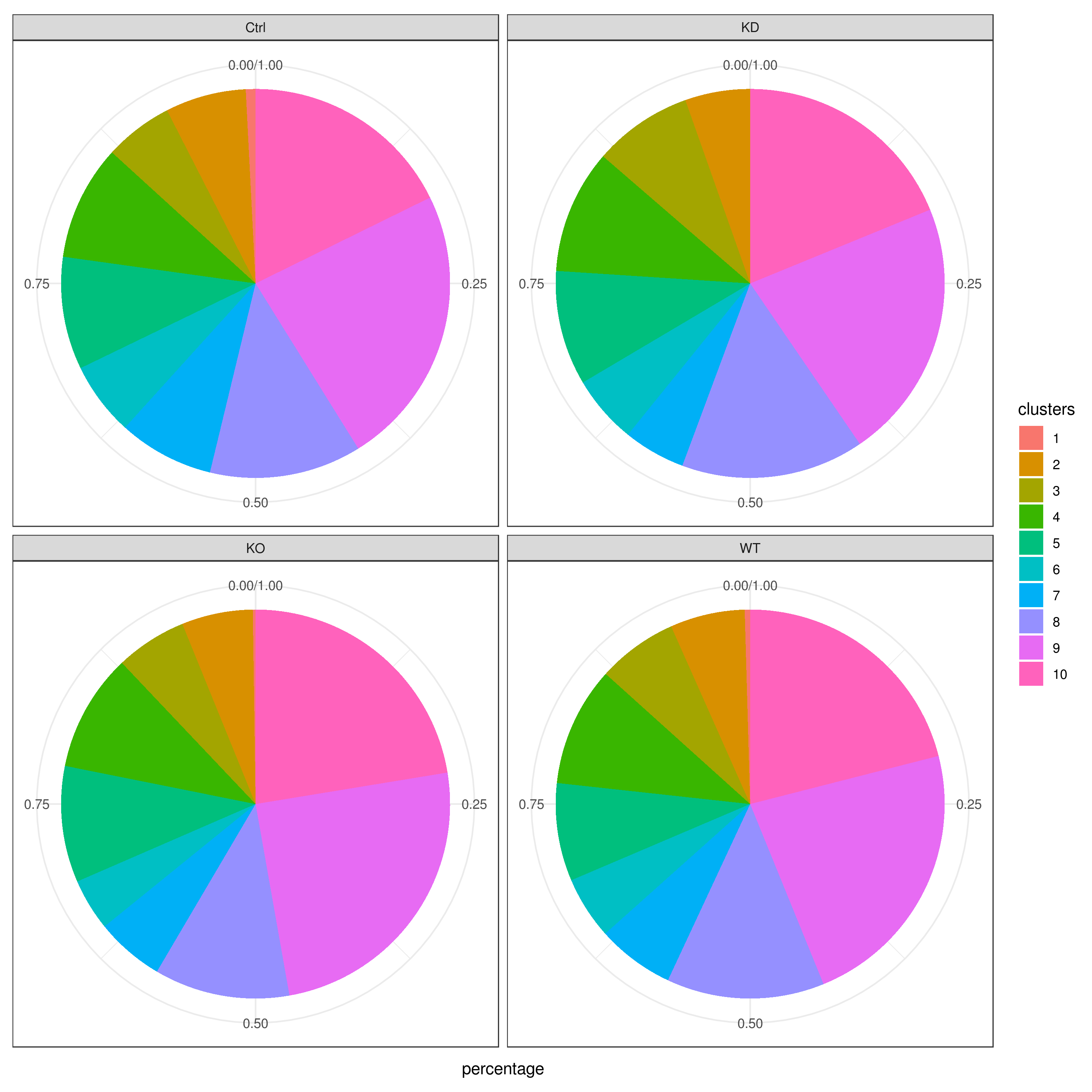
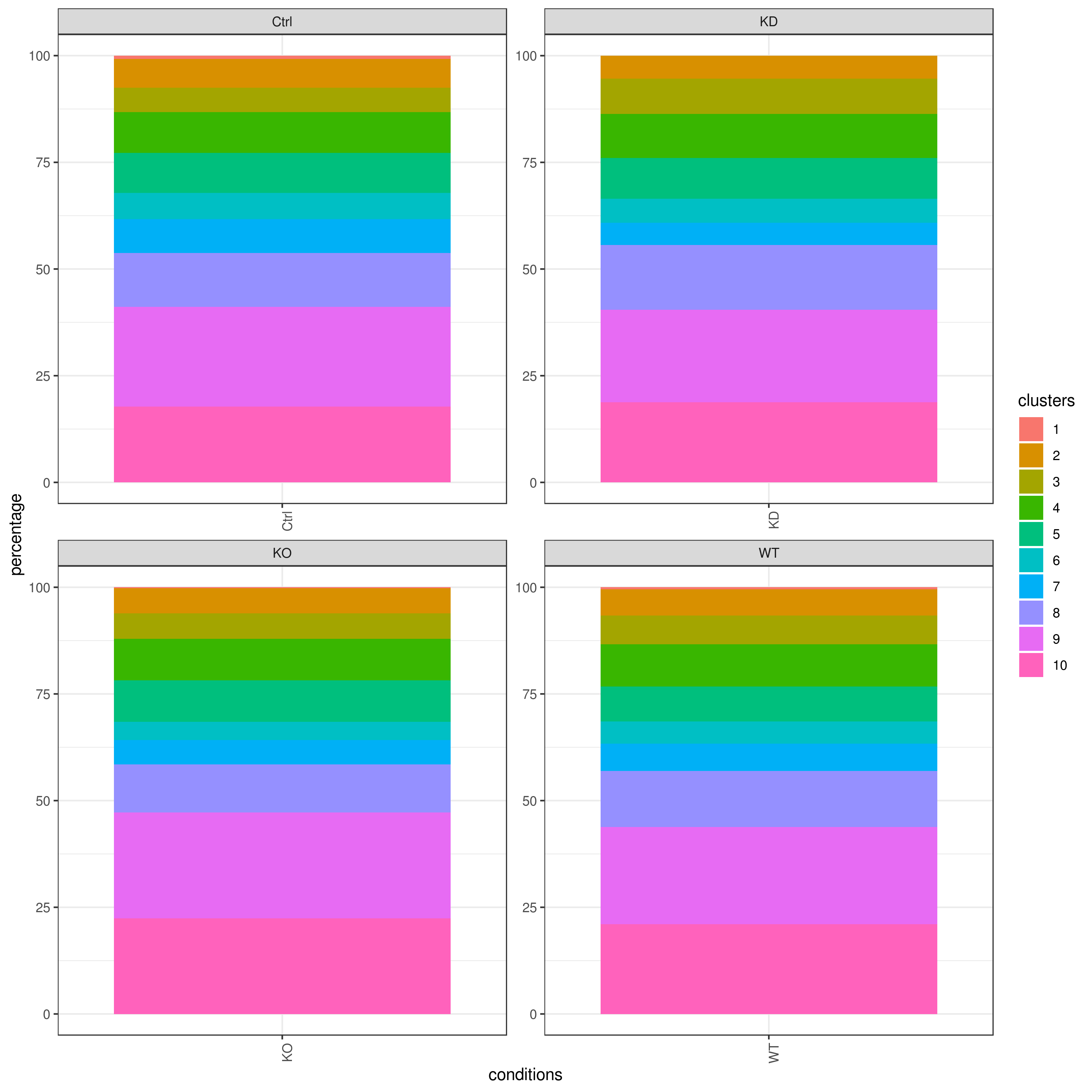
- Cell frequencies and proportions
clust.cond.info(my.obj, plot.type = "pie", normalize.ncell = TRUE, my.out.put = "plot", normalize.by = "percentage")
clust.cond.info(my.obj, plot.type = "bar", normalize.ncell = TRUE,my.out.put = "plot", normalize.by = "percentage")
clust.cond.info(my.obj, plot.type = "pie.cond", normalize.ncell = T, my.out.put = "plot", normalize.by = "percentage")
clust.cond.info(my.obj, plot.type = "bar.cond", normalize.ncell = T,my.out.put = "plot", normalize.by = "percentage")
my.obj <- clust.cond.info(my.obj)
head(my.obj@my.freq)
# conditions TC SF clusters Freq Norm.Freq percentage
#1 Ctrl 491 1.265 1 4 3.162 0.81
#2 Ctrl 491 1.265 11 32 25.296 6.52
#3 Ctrl 491 1.265 8 114 90.119 23.22
#4 Ctrl 491 1.265 5 43 33.992 8.76
#5 Ctrl 491 1.265 2 33 26.087 6.72
#6 Ctrl 491 1.265 9 86 67.984 17.52
- Cluster QC
clust.stats.plot(my.obj, plot.type = "box.mito", interactive = F)
clust.stats.plot(my.obj, plot.type = "box.gene", interactive = F)
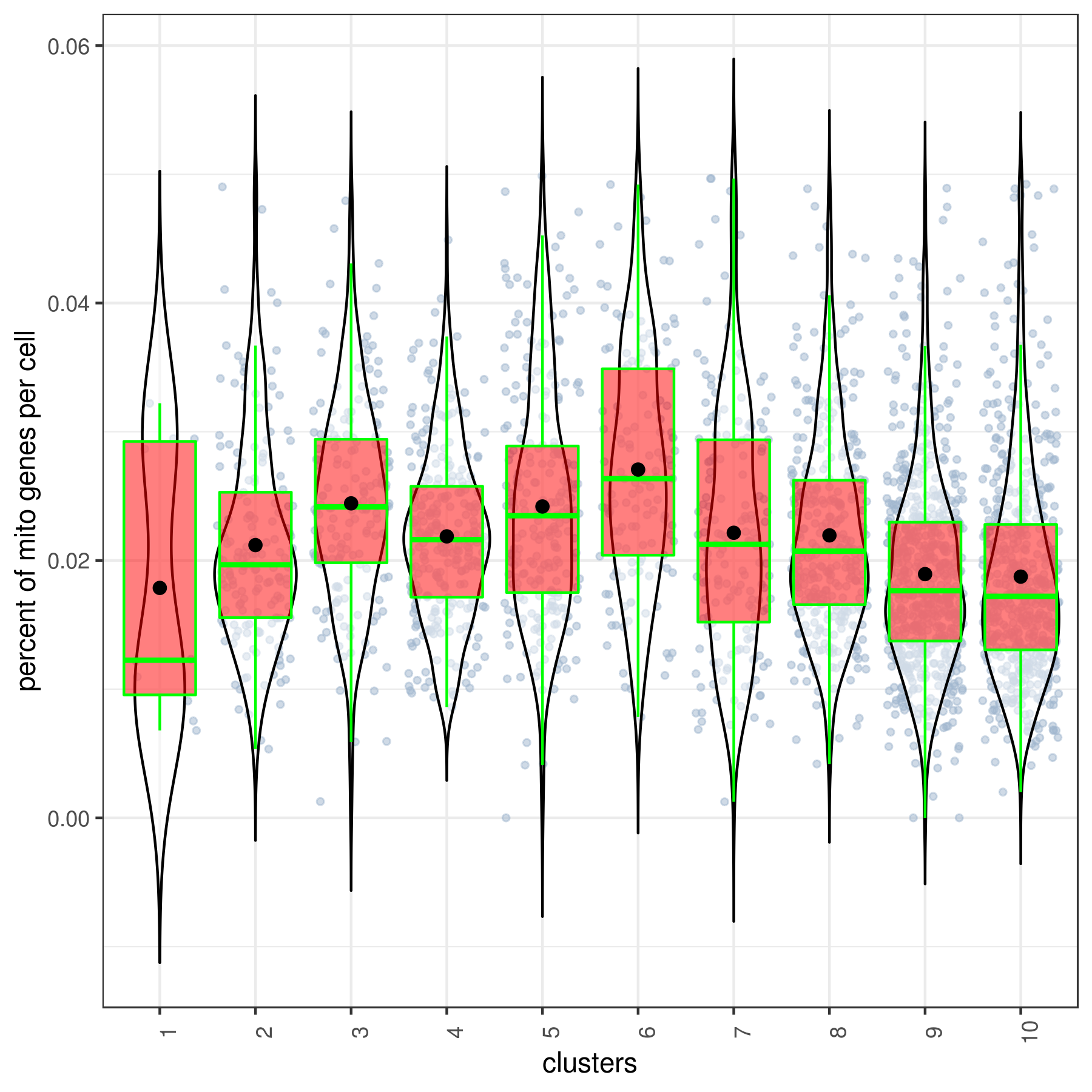
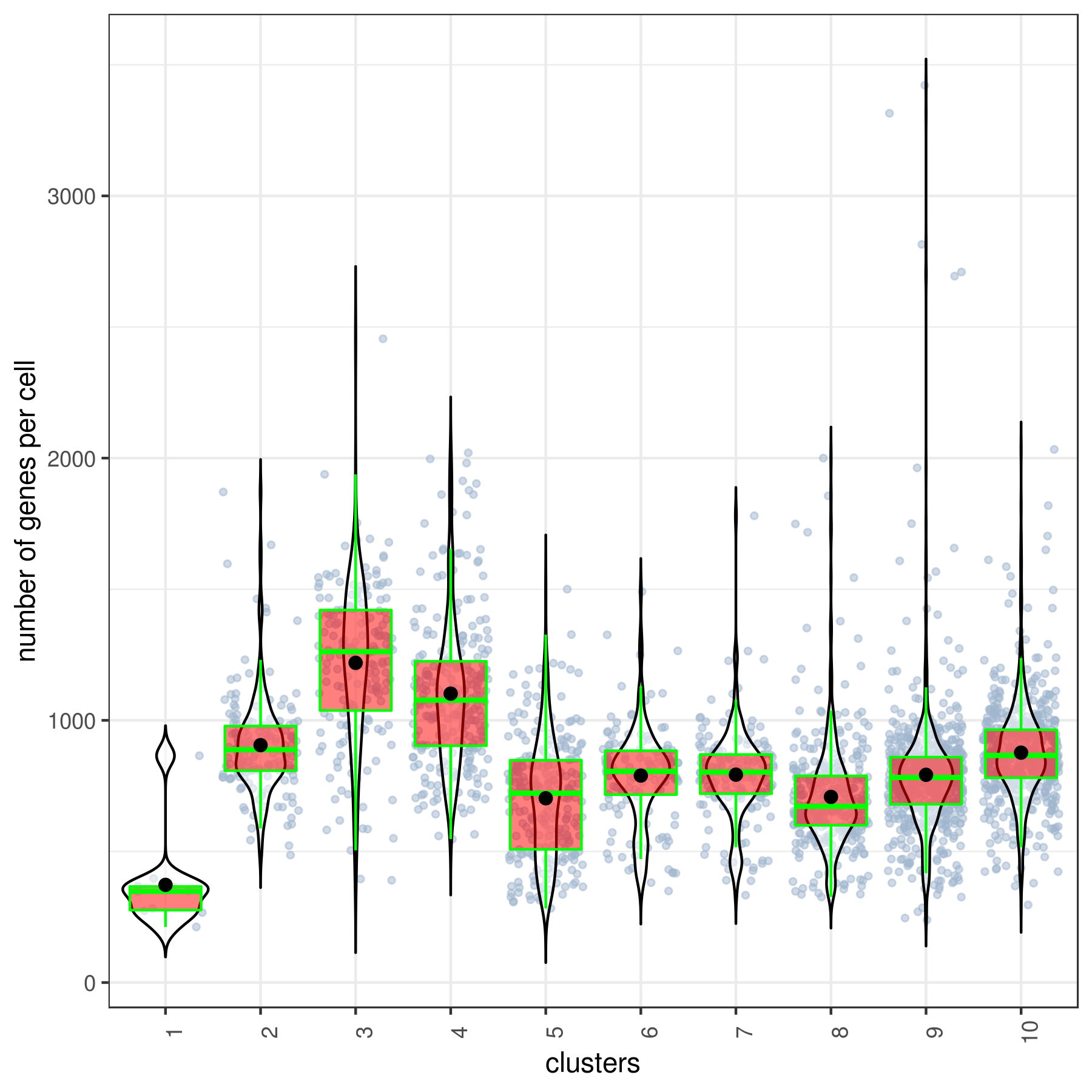
- Run data imputation
my.obj <- run.impute(my.obj, dims = 1:10, nn = 10, data.type = "pca")- Save your object
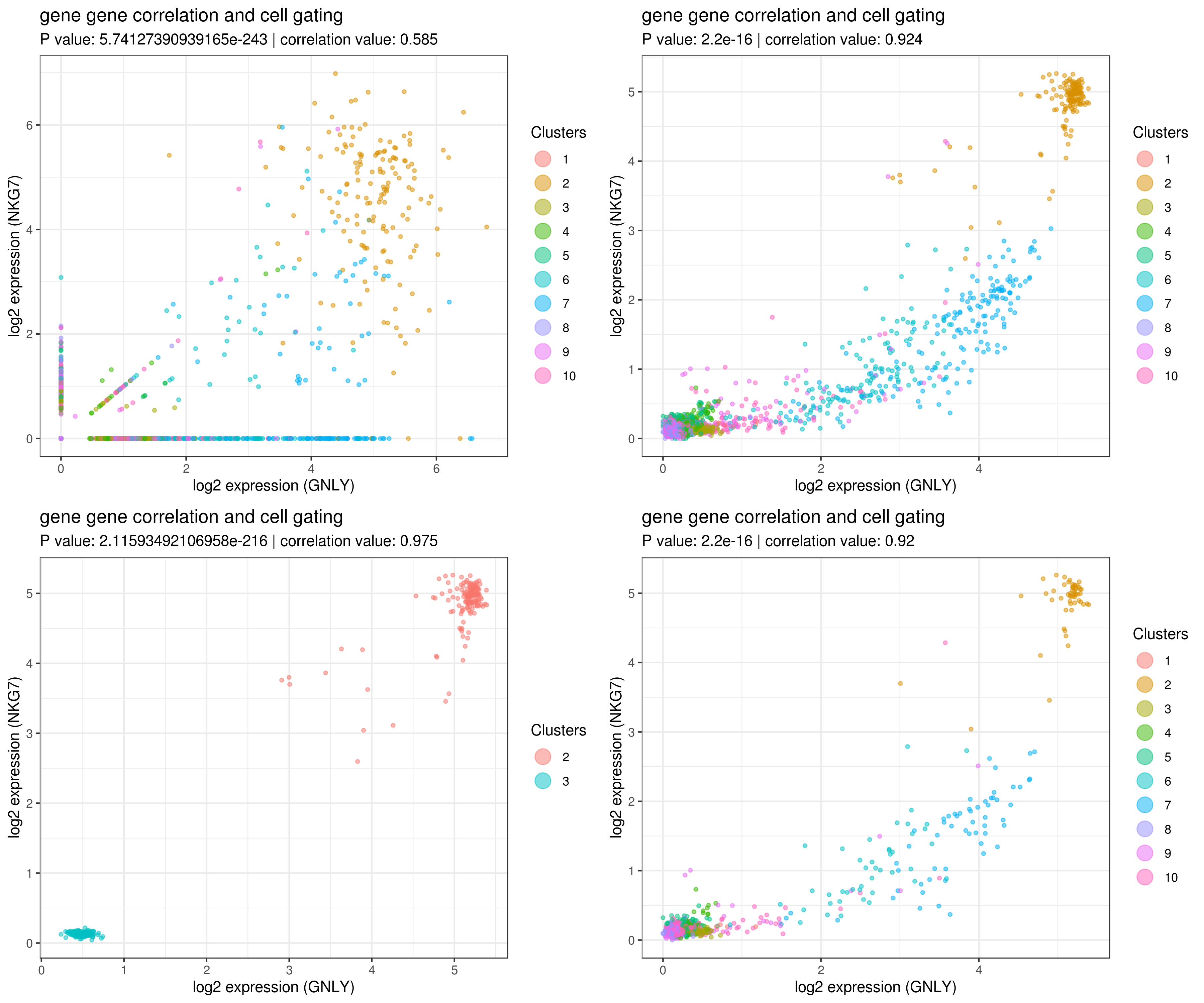
save(my.obj, file = "my.obj.Robj")- gene gene correlation
# impute more cells by increasing nn for better resulst.
my.obj <- run.impute(my.obj,dims = 1:10,data.type = "pca", nn = 50)
# main data
A <- gg.cor(my.obj,
interactive = F,
gene1 = "GNLY",
gene2 = "NKG7",
conds = NULL,
clusts = NULL,
data.type = "main")
# imputed data
B <- gg.cor(my.obj,
interactive = F,
gene1 = "GNLY",
gene2 = "NKG7",
conds = NULL,
clusts = NULL,
data.type = "imputed")
C <- gg.cor(my.obj,
interactive = F,
gene1 = "GNLY",
gene2 = "NKG7",
conds = NULL,
clusts = c(3,2),
data.type = "imputed")
# imputed data
D <- gg.cor(my.obj,
interactive = F,
gene1 = "GNLY",
gene2 = "NKG7",
conds = c("WT"),
clusts = NULL,
data.type = "imputed")
grid.arrange(A,B,C,D)
- Find marker genes
marker.genes <- findMarkers(my.obj,
fold.change = 2,
padjval = 0.1)
dim(marker.genes)
# [1] 1070 17
head(marker.genes)
# baseMean baseSD AvExpInCluster AvExpInOtherClusters foldChange
#PPBP 0.8257760 12.144694 181.3945 0.1399852 1295.8120969
#GPX1 1.3989591 4.344717 57.4034 1.1862571 48.3903523
#CALM3 0.5469743 1.230942 10.7848 0.5080915 21.2260968
#OAZ1 4.9077851 5.979586 46.7867 4.7487311 9.8524635
#MYL6 3.0806167 3.562124 21.3690 3.0111584 7.0966045
#CD74 8.5523704 13.359205 2.6120 8.5749316 0.3046088
# log2FoldChange pval padj clusters gene cluster_1
#PPBP 10.339641 1.586683e-06 0.014786300 1 PPBP 181.3945
#GPX1 5.596648 1.107541e-07 0.001103775 1 GPX1 57.4034
#CALM3 4.407767 2.098341e-06 0.019415953 1 CALM3 10.7848
#OAZ1 3.300485 7.857814e-07 0.007464137 1 OAZ1 46.7867
#MYL6 2.827129 1.296112e-06 0.012156230 1 MYL6 21.3690
#CD74 -1.714970 9.505749e-06 0.083983296 1 CD74 2.6120
# cluster_2 cluster_3 cluster_4 cluster_5 cluster_6 cluster_7
#PPBP 0.0000000 0.1444327 0.2282912 0.0640625 0.01739706 0.1541084
#GPX1 0.2424969 1.2218772 3.9292720 4.4329583 0.25663235 0.2712831
#CALM3 0.6537205 0.8149415 0.6071034 0.5245625 0.44687500 0.5081867
#OAZ1 3.2077826 12.2072339 8.6080077 10.8738208 2.71288971 3.6402289
#MYL6 4.9660870 5.7945673 4.2813218 4.3046458 2.42854412 3.9030542
#CD74 2.9385839 8.9848538 15.7646245 5.9454250 2.19555882 3.8323072
# cluster_8 cluster_9 cluster_10
#PPBP 0.02478274 0.3668433 0.01026335
#GPX1 0.61210714 0.4635153 0.39311786
#CALM3 0.22591369 0.5210339 0.48856538
#OAZ1 3.67225595 2.3590420 2.53362063
#MYL6 1.72344048 1.6460420 2.59901289
#CD74 36.10877976 1.5638853 1.82587477
# baseMean: average expression in all the cells
# baseSD: Standard Deviation
# AvExpInCluster: average expression in cluster number (see clusters)
# AvExpInOtherClusters: average expression in all the other clusters
# foldChange: AvExpInCluster/AvExpInOtherClusters
# log2FoldChange: log2(AvExpInCluster/AvExpInOtherClusters)
# pval: P value
# padj: Adjusted P value
# clusters: marker for cluster number
# gene: marker gene for the cluster
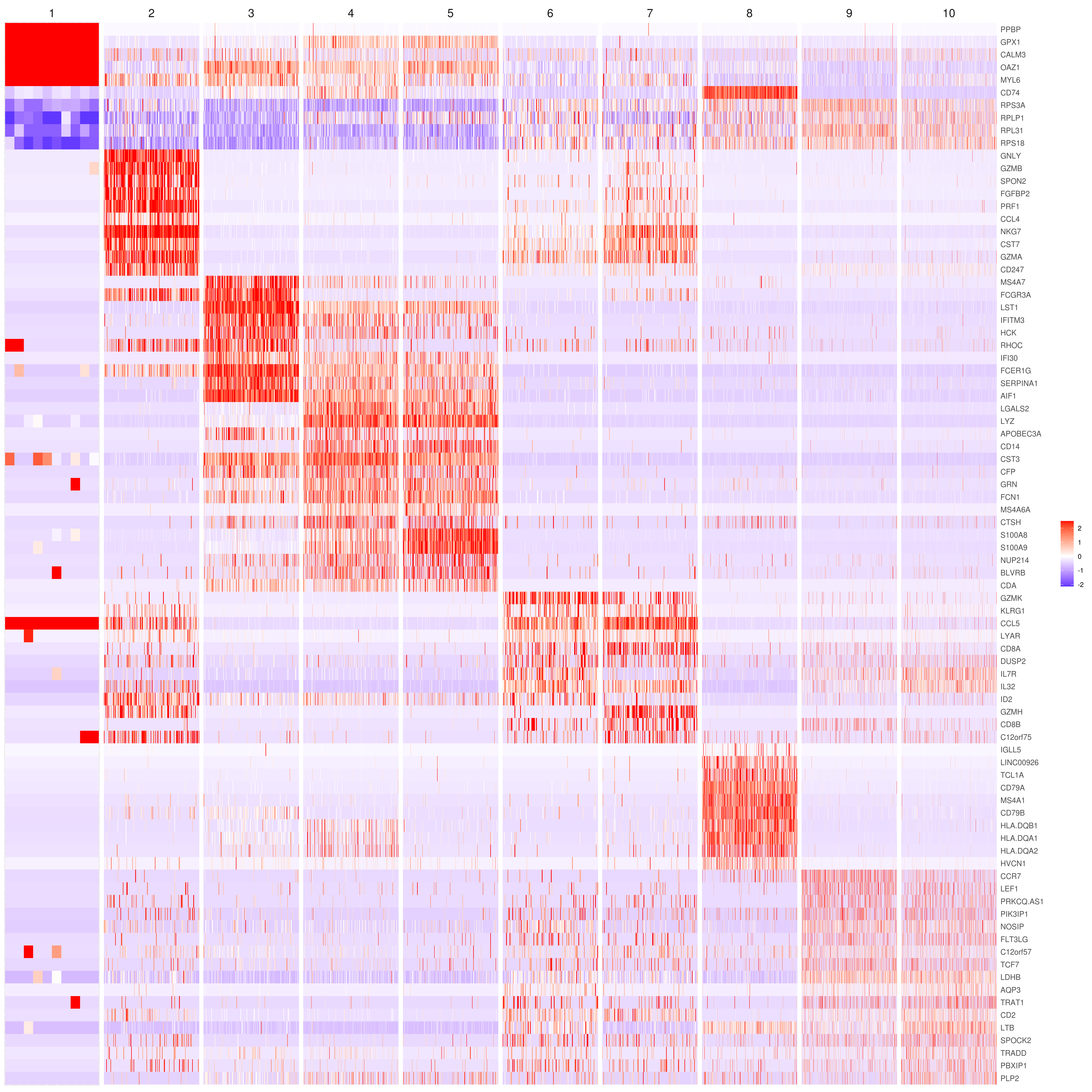
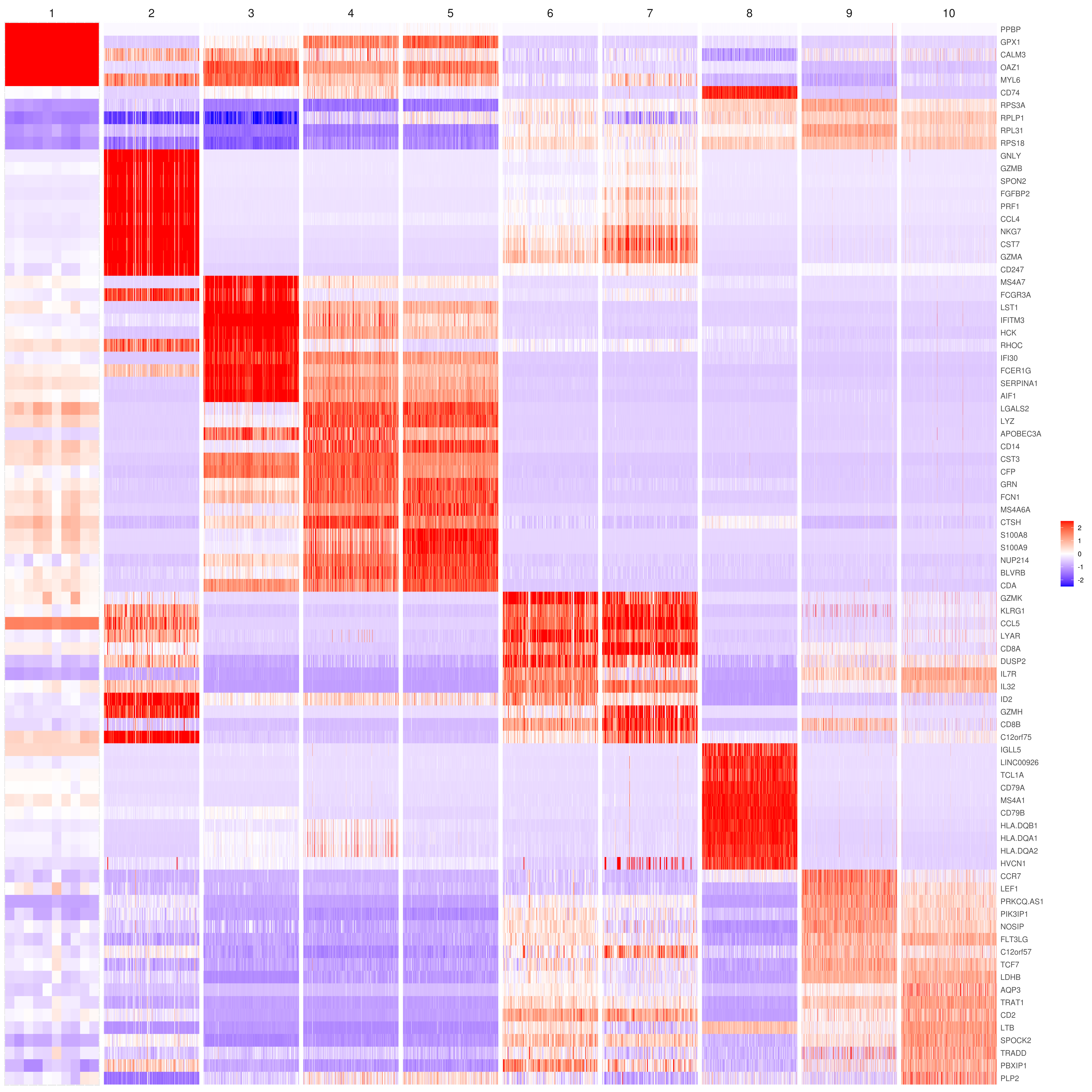
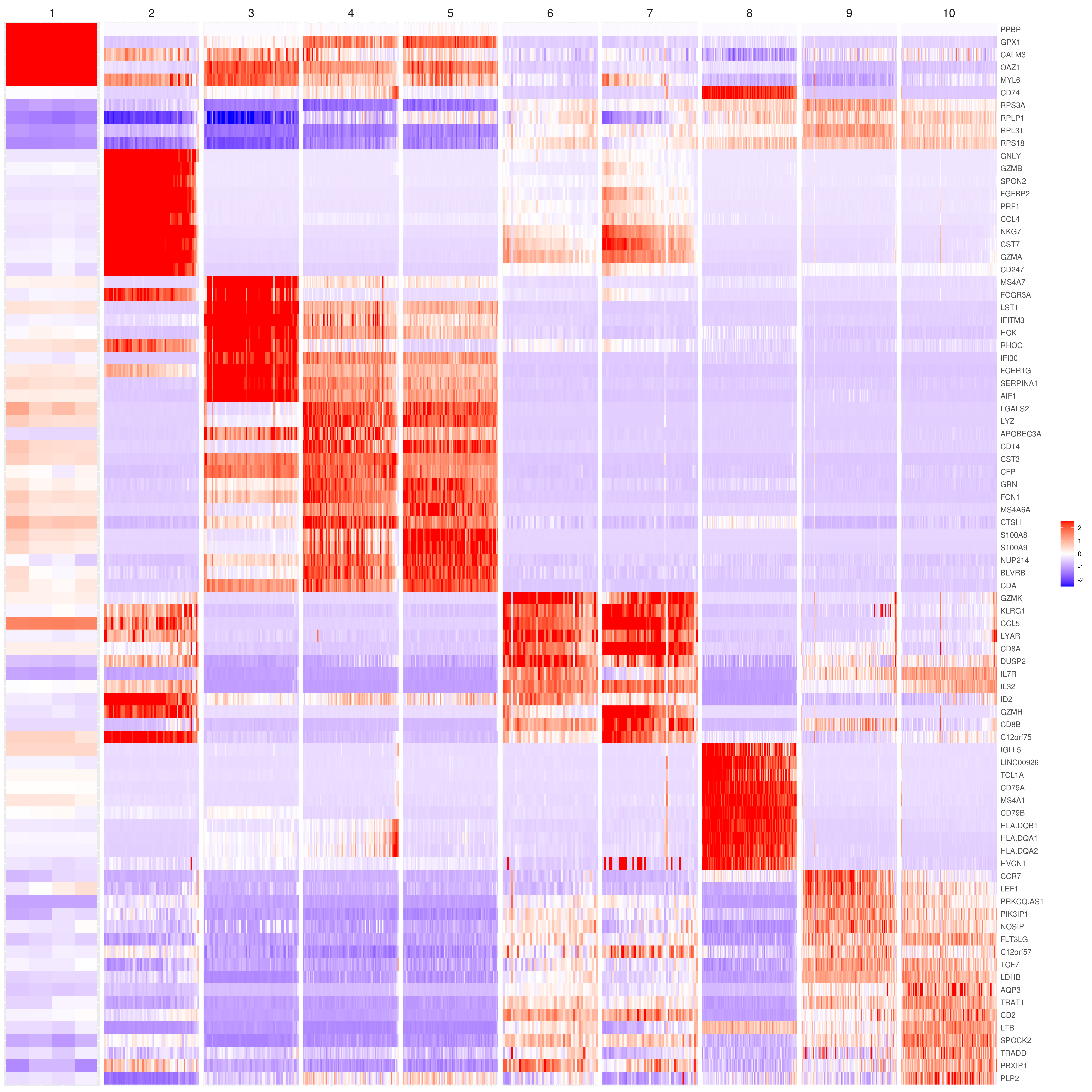
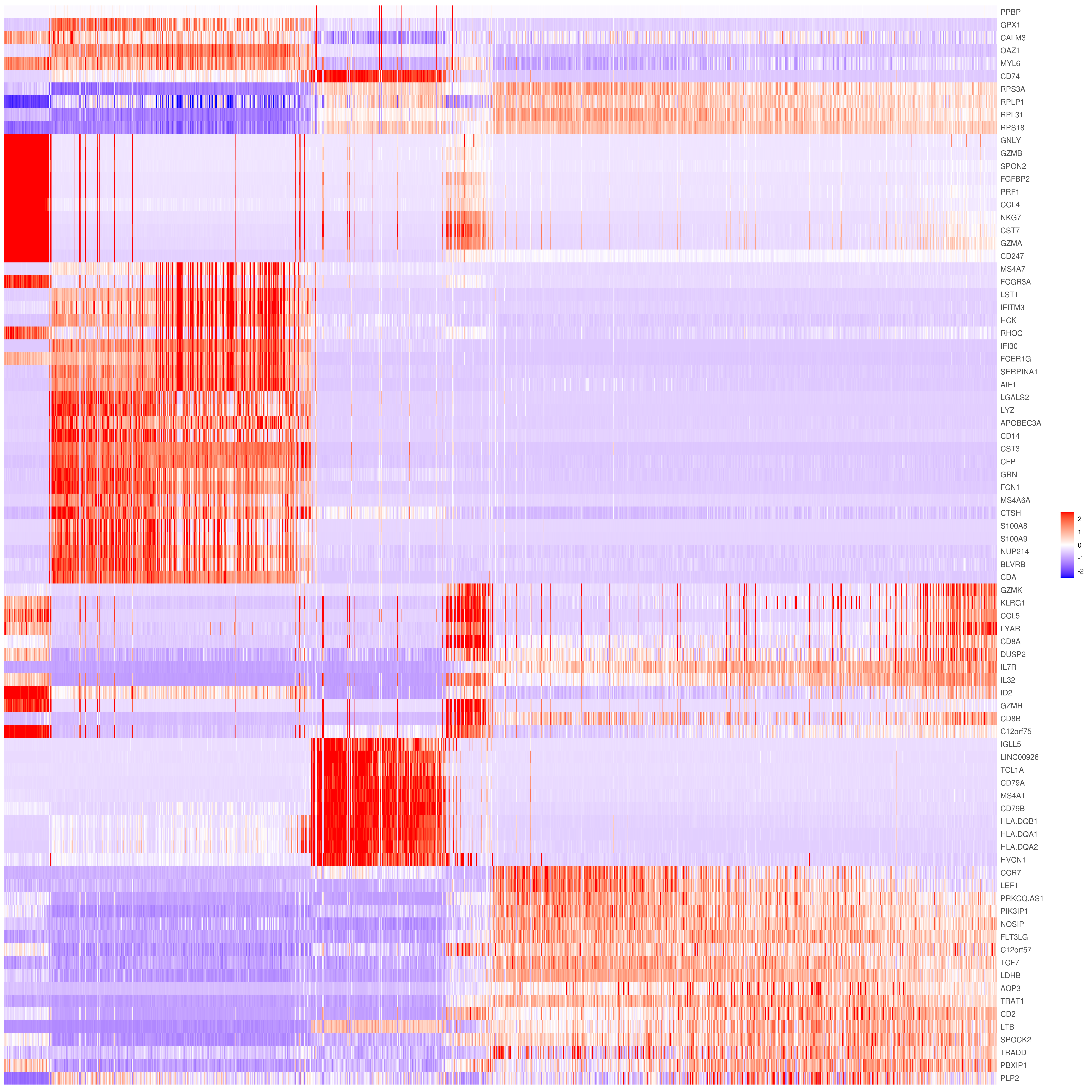
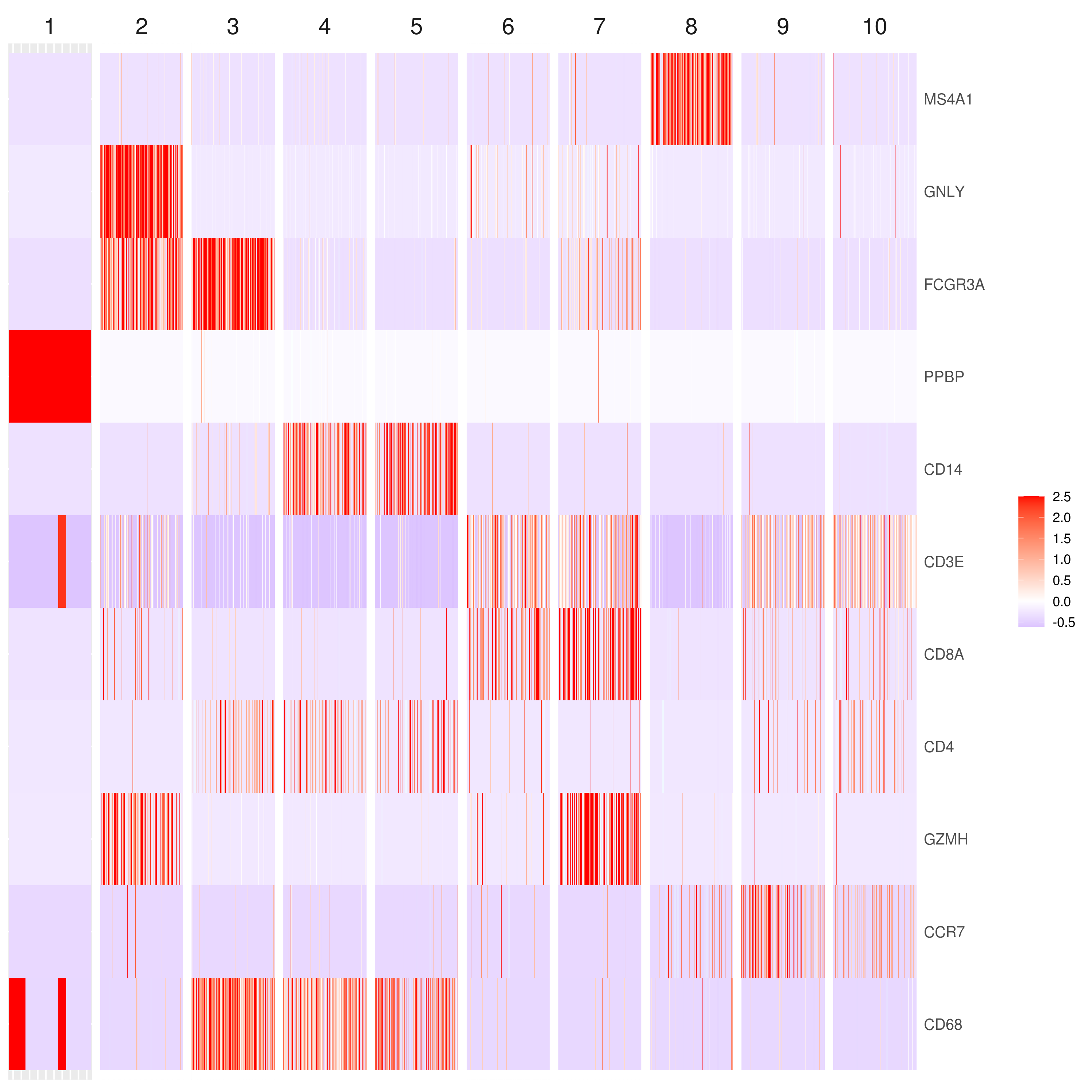
# the rest are the average expression for each cluster- Heatmap
# find top genes
MyGenes <- top.markers(marker.genes, topde = 10, min.base.mean = 0.2,filt.ambig = F)
MyGenes <- unique(MyGenes)
# main data
heatmap.gg.plot(my.obj, gene = MyGenes, interactive = F, cluster.by = "clusters", conds.to.plot = NULL)
# imputed data
heatmap.gg.plot(my.obj, gene = MyGenes, interactive = F, cluster.by = "clusters", data.type = "imputed", conds.to.plot = NULL)
# sort cells and plot only one condition
heatmap.gg.plot(my.obj, gene = MyGenes, interactive = F, cluster.by = "clusters", data.type = "imputed", cell.sort = TRUE, conds.to.plot = c("WT"))
# Pseudotime stile
heatmap.gg.plot(my.obj, gene = MyGenes, interactive = F, cluster.by = "none", data.type = "imputed", cell.sort = TRUE)
# intractive
# heatmap.gg.plot(my.obj, gene = MyGenes, interactive = T, out.name = "heatmap_gg", cluster.by = "clusters")
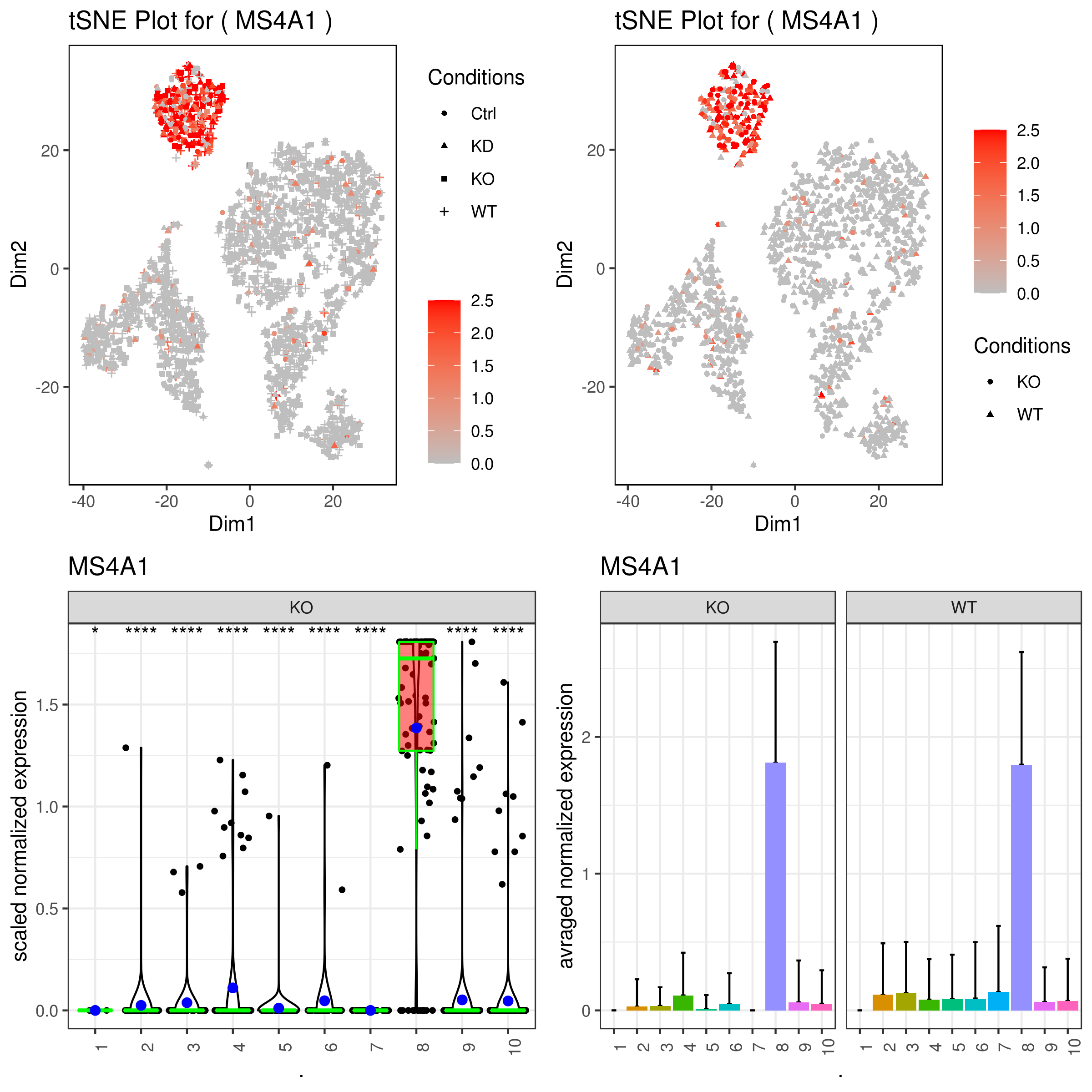
- Plot genes
A <- gene.plot(my.obj, gene = "MS4A1",
plot.type = "scatterplot",
interactive = F,
out.name = "scatter_plot")
# PCA 2D
B <- gene.plot(my.obj, gene = "MS4A1",
plot.type = "scatterplot",
interactive = F,
out.name = "scatter_plot",
plot.data.type = "umap")
# Box Plot
C <- gene.plot(my.obj, gene = "MS4A1",
box.to.test = 0,
box.pval = "sig.signs",
col.by = "clusters",
plot.type = "boxplot",
interactive = F,
out.name = "box_plot")
# Bar plot (to visualize fold changes)
D <- gene.plot(my.obj, gene = "MS4A1",
col.by = "clusters",
plot.type = "barplot",
interactive = F,
out.name = "bar_plot")
library(gridExtra)
png('gene.plots.png', width = 8, height = 8, units = 'in', res = 300)
grid.arrange(A,B,C,D)
dev.off()
### same on imputed data
A <- gene.plot(my.obj, gene = "MS4A1",
plot.type = "scatterplot",
interactive = F,
data.type = "imputed",
out.name = "scatter_plot")
# PCA 2D
B <- gene.plot(my.obj, gene = "MS4A1",
plot.type = "scatterplot",
interactive = F,
out.name = "scatter_plot",
data.type = "imputed",
plot.data.type = "umap")
# Box Plot
C <- gene.plot(my.obj, gene = "MS4A1",
box.to.test = 0,
box.pval = "sig.signs",
col.by = "clusters",
plot.type = "boxplot",
interactive = F,
data.type = "imputed",
out.name = "box_plot")
# Bar plot (to visualize fold changes)
D <- gene.plot(my.obj, gene = "MS4A1",
col.by = "clusters",
plot.type = "barplot",
interactive = F,
data.type = "imputed",
out.name = "bar_plot")
library(gridExtra)
png('gene.plots_imputed.png', width = 8, height = 8, units = 'in', res = 300)
grid.arrange(A,B,C,D)
dev.off()
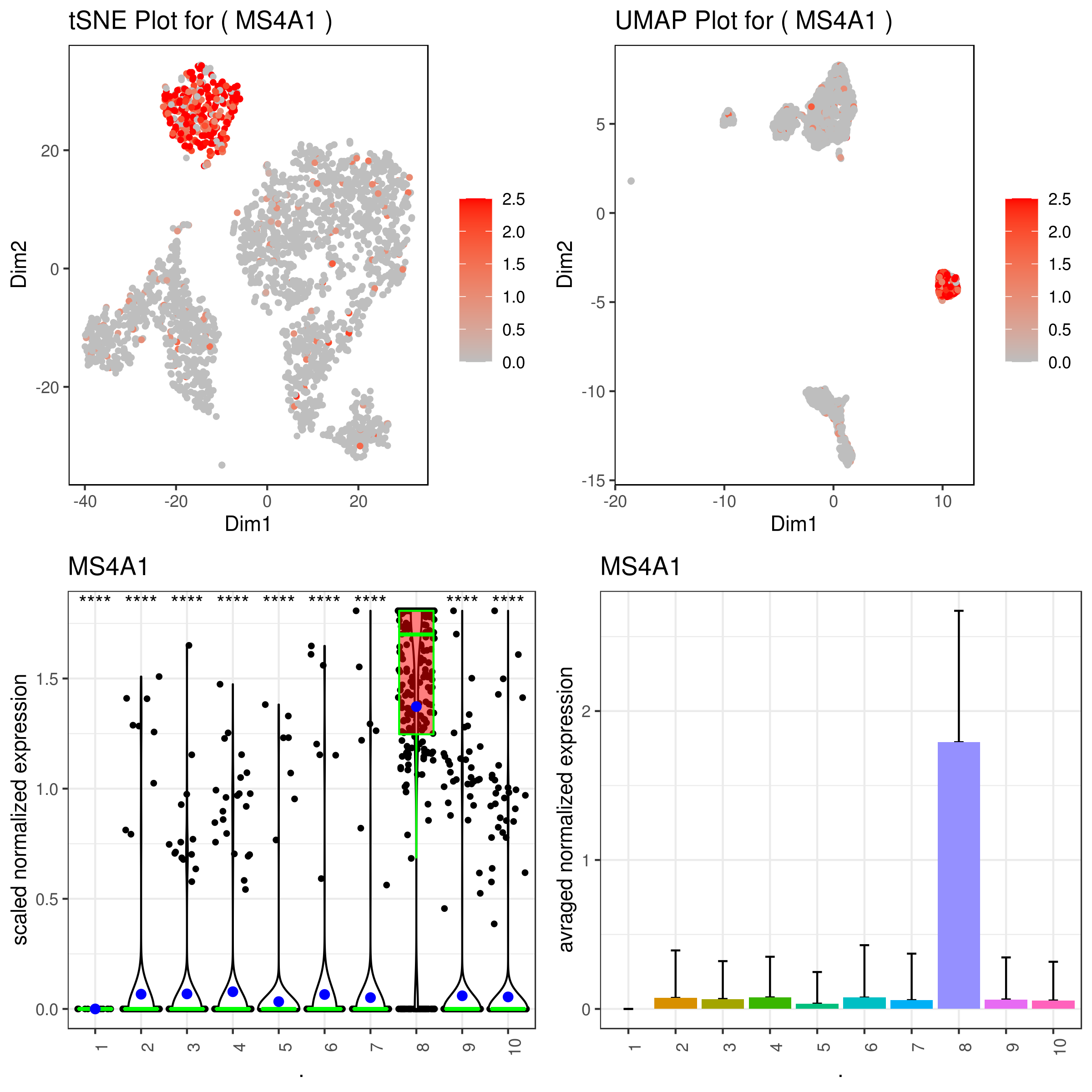
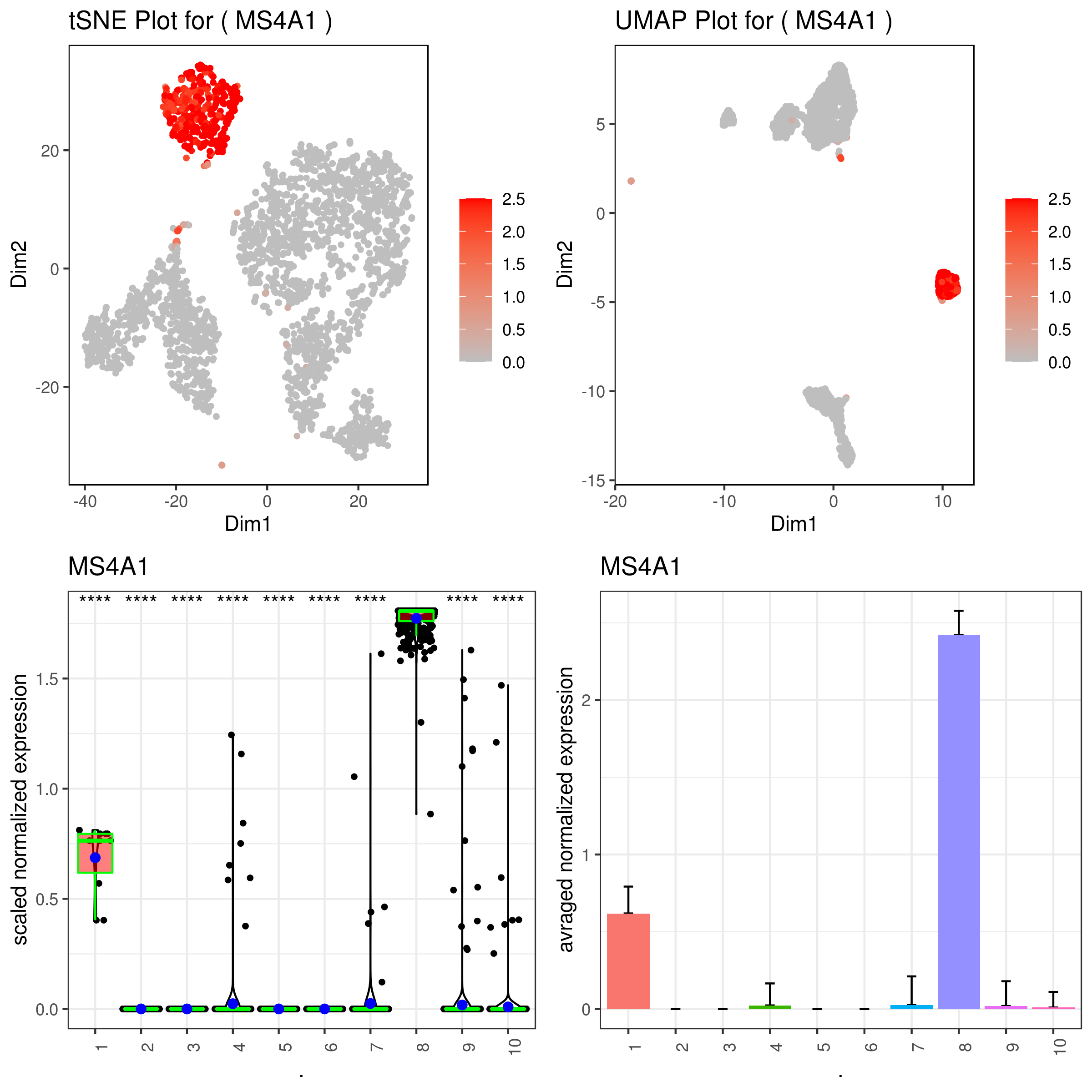
- Multiple plots
Change the section in between #### signs for different plots (e.g. boxplot, bar, ...).
genelist = c("MS4A1","GNLY","FCGR3A","NKG7","CD14","CD3E","CD8A","CD4","GZMH","CCR7","CD68")
rm(list = ls(pattern="PL_"))
for(i in genelist){
####
MyPlot <- gene.plot(my.obj, gene = i,
interactive = F,
cell.size = 0.1,
plot.data.type = "knetl",
data.type = "main",
scaleValue = T,
min.scale = 0,max.scale = 2.0,
cell.transparency = 1)
####
NameCol=paste("PL",i,sep="_")
eval(call("<-", as.name(NameCol), MyPlot))
}
library(cowplot)
filenames <- ls(pattern="PL_")
B <- cluster.plot(my.obj,plot.type = "knetl",interactive = F,cell.size = 0.1,cell.transparency = 1,anno.clust=T)
filenames <- c("B",filenames)
png('genes_KNetL.png',width = 15, height = 12, units = 'in', res = 300)
plot_grid(plotlist=mget(filenames))
dev.off()
# or heatmap
# heatmap.gg.plot(my.obj, gene = genelist, interactive = F, cluster.by = "clusters")
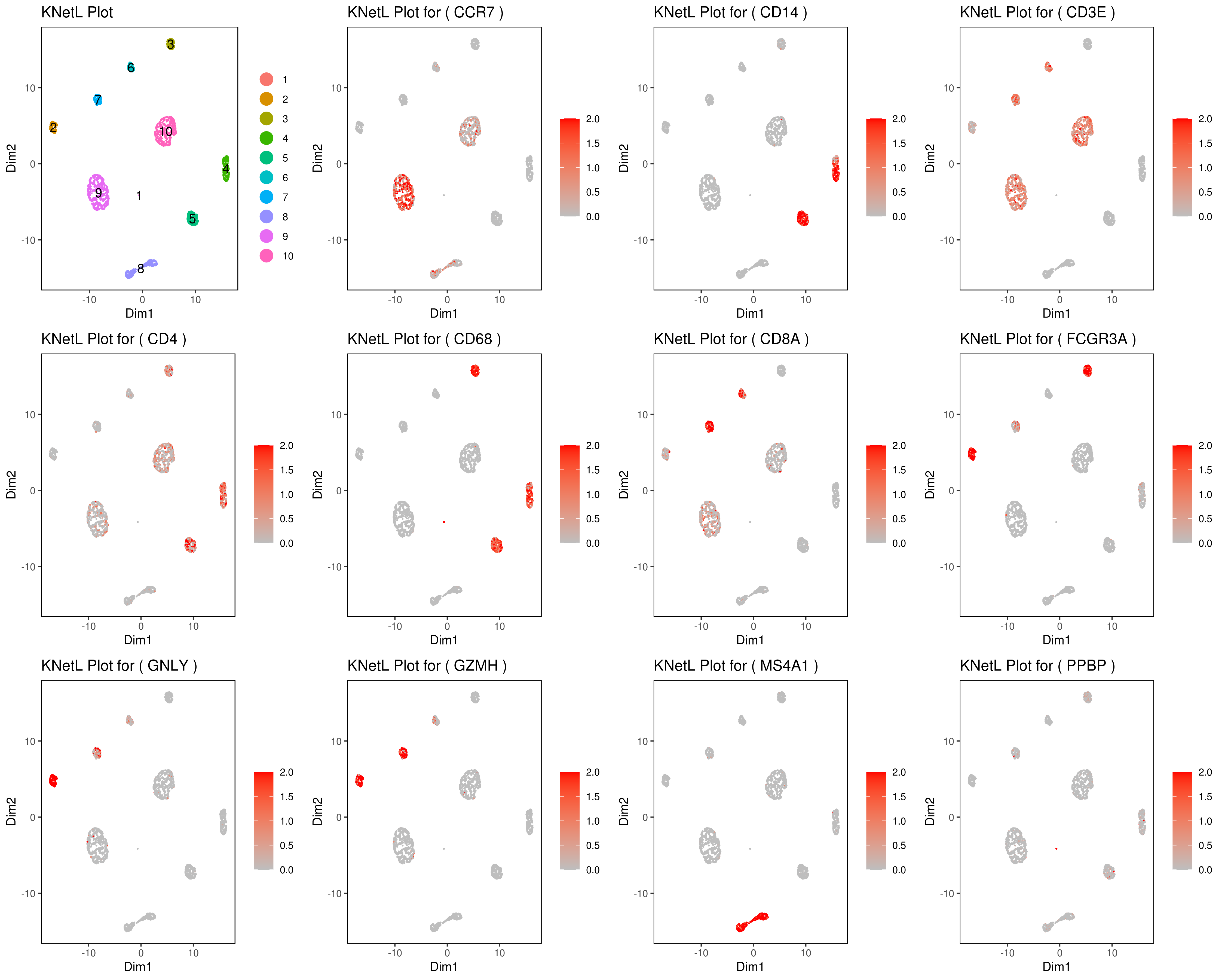
- Make your own customized plots
# You can export the data using this command (one or multiple genes):
gene.plot(my.obj, gene = "MS4A1", write.data = T, scaleValue = F, data.type = "main")
# This would create a text file called "MS4A1.tsv".
head(read.table("MS4A1.tsv"))
# V1 V2 Expression Clusters Conditions
#WT_AAACATACAACCAC.1 12.499481 -11.436633 0.000000 9 WT
#WT_AAACATTGAGCTAC.1 -8.783793 24.417999 1.942233 8 WT
#WT_AAACATTGATCAGC.1 -2.650761 10.932273 0.000000 10 WT
#WT_AAACCGTGCTTCCG.1 -28.916702 -5.542731 0.000000 4 WT
#WT_AAACCGTGTATGCG.1 21.211557 -31.626822 0.000000 2 WT
#WT_AAACGCACTGGTAC.1 5.225419 -5.141192 0.000000 10 WT
# you use this to make your own plots in ggplot2 or other visualization packages. - Annotating clusters
###### Labeling the clusters
#CD3E: only in T Cells
#FCGR3A (CD16): in CD16+ monocytes and some expression NK cells
#GNLY: NK cells
#MS4A1: B cells
#GZMH: in GZMH+ T8 cells and some expression NK cells
#CD8A: in T8 cells
#CD4: in T4 and some myeloid cells
#CCR7: expressed more in memory cells
#CD14: in CD14+ monocytes
#CD68: in monocytes/MF
my.obj <- change.clust(my.obj, change.clust = 1, to.clust = "001.MG")
my.obj <- change.clust(my.obj, change.clust = 2, to.clust = "002.NK")
my.obj <- change.clust(my.obj, change.clust = 3, to.clust = "003.CD16+.Mono")
my.obj <- change.clust(my.obj, change.clust = 4, to.clust = "004.MF")
my.obj <- change.clust(my.obj, change.clust = 5, to.clust = "005.CD14+.Mono")
my.obj <- change.clust(my.obj, change.clust = 6, to.clust = "006.Naive.T8")
my.obj <- change.clust(my.obj, change.clust = 7, to.clust = "007.GZMH+.T8")
my.obj <- change.clust(my.obj, change.clust = 8, to.clust = "008.B")
my.obj <- change.clust(my.obj, change.clust = 9, to.clust = "009.Memory.T4")
my.obj <- change.clust(my.obj, change.clust = 10, to.clust = "010.Naive.T4")
A= cluster.plot(my.obj,plot.type = "pca",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
B= cluster.plot(my.obj,plot.type = "umap",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=T)
C= cluster.plot(my.obj,plot.type = "tsne",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=T)
D= cluster.plot(my.obj,plot.type = "knetl",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=T)
grid.arrange(A,B,C,D)
- Plotting conditions and clusters for genes
A <- gene.plot(my.obj, gene = "MS4A1",
plot.type = "scatterplot",
interactive = F,
cell.transparency = 1,
scaleValue = TRUE,
min.scale = 0,
max.scale = 2.5,
back.col = "white",
cond.shape = TRUE)
B <- gene.plot(my.obj, gene = "MS4A1",
plot.type = "scatterplot",
interactive = F,
cell.transparency = 1,
scaleValue = TRUE,
min.scale = 0,
max.scale = 2.5,
back.col = "white",
cond.shape = TRUE,
conds.to.plot = c("KO","WT"))
C <- gene.plot(my.obj, gene = "MS4A1",
plot.type = "boxplot",
interactive = F,
back.col = "white",
cond.shape = TRUE,
conds.to.plot = c("KO"))
D <- gene.plot(my.obj, gene = "MS4A1",
plot.type = "barplot",
interactive = F,
cell.transparency = 1,
back.col = "white",
cond.shape = TRUE,
conds.to.plot = c("KO","WT"))
library(gridExtra)
grid.arrange(A,B,C,D)
- Some example 2D and 3D plots and plotting clusters and conditions at the same time
# example
cluster.plot(my.obj,
cell.size = 1,
plot.type = "umap",
cell.color = "black",
back.col = "white",
col.by = "clusters",
cell.transparency = 0.5,
clust.dim = 2,
cond.shape = T,
interactive = T,
out.name = "2d_UMAP_clusters_conds")
# 2D
cluster.plot(my.obj,
cell.size = 1,
plot.type = "tsne",
cell.color = "black",
back.col = "white",
col.by = "clusters",
cell.transparency = 0.5,
clust.dim = 2,
interactive = F)
# interactive 2D
cluster.plot(my.obj,
plot.type = "tsne",
col.by = "clusters",
clust.dim = 2,
interactive = T,
out.name = "tSNE_2D_clusters")
# interactive 3D
cluster.plot(my.obj,
plot.type = "tsne",
col.by = "clusters",
clust.dim = 3,
interactive = T,
out.name = "tSNE_3D_clusters")
# Density plot for clusters
cluster.plot(my.obj,
plot.type = "pca",
col.by = "clusters",
interactive = F,
density=T)
# Density plot for conditions
cluster.plot(my.obj,
plot.type = "pca",
col.by = "conditions",
interactive = F,
density=T)
cluster.plot(my.obj,
cell.size = 1,
plot.type = "diffusion",
cell.color = "black",
back.col = "white",
col.by = "clusters",
cell.transparency = 0.5,
clust.dim = 2,
interactive = F)
cluster.plot(my.obj,
cell.size = 1,
plot.type = "diffusion",
cell.color = "black",
back.col = "white",
col.by = "clusters",
cell.transparency = 0.5,
clust.dim = 3,
interactive = F)







- Differential Expression Analysis
The differential expression (DE) analysis function in iCellR allows the users to choose from any combinations of clusters and conditions. For example, a user with two samples (say WT and KO) has four different possible ways of comparisons:
a-Comparing a cluster/clusters with different cluster/clusters (e.g. cluster 1 and 2 vs. 4)
b-Comparing a cluster/clusters with different cluster/clusters only in one/more condition/conditions (e.g. cluster 1 vs cluster 2 but only the WT sample)
c-Comparing a condition/conditions with different condition/conditions (e.g. WT vs KO)
d-Comparing a condition/conditions with different condition/conditions only in one/more cluster/clusters (e.g. cluster 1 WT vs cluster 1 KO)
diff.res <- run.diff.exp(my.obj, de.by = "clusters", cond.1 = c(1,4), cond.2 = c(2))
diff.res1 <- as.data.frame(diff.res)
diff.res1 <- subset(diff.res1, padj < 0.05)
head(diff.res1)
# baseMean 1_4 2 foldChange log2FoldChange pval
#AAK1 0.19554589 0.26338228 0.041792762 0.15867719 -2.655833 8.497012e-33
#ABHD14A 0.09645732 0.12708519 0.027038379 0.21275791 -2.232715 1.151865e-11
#ABHD14B 0.19132829 0.23177944 0.099644572 0.42991118 -1.217889 3.163623e-09
#ABLIM1 0.06901900 0.08749258 0.027148089 0.31029018 -1.688310 1.076382e-06
#AC013264.2 0.07383608 0.10584821 0.001279649 0.01208947 -6.370105 1.291674e-19
#AC092580.4 0.03730859 0.05112053 0.006003441 0.11743700 -3.090041 5.048838e-07
padj
#AAK1 1.294690e-28
#ABHD14A 1.708446e-07
#ABHD14B 4.636290e-05
#ABLIM1 1.540087e-02
#AC013264.2 1.950557e-15
#AC092580.4 7.254675e-03
# more examples
# Comparing a condition/conditions with different condition/conditions (e.g. WT vs KO)
diff.res <- run.diff.exp(my.obj, de.by = "conditions", cond.1 = c("WT"), cond.2 = c("KO"))
# Comparing a cluster/clusters with different cluster/clusters (e.g. cluster 1 and 2 vs. 4)
diff.res <- run.diff.exp(my.obj, de.by = "clusters", cond.1 = c(1,4), cond.2 = c(2))
# Comparing a condition/conditions with different condition/conditions only in one/more cluster/clusters (e.g. cluster 1 WT vs cluster 1 KO)
diff.res <- run.diff.exp(my.obj, de.by = "clustBase.condComp", cond.1 = c("WT"), cond.2 = c("KO"), base.cond = 1)
# Comparing a cluster/clusters with different cluster/clusters only in one/more condition/conditions (e.g. cluster 1 vs cluster 2 but only the WT sample)
diff.res <- run.diff.exp(my.obj, de.by = "condBase.clustComp", cond.1 = c(1), cond.2 = c(2), base.cond = "WT")- Volcano and MA plots
# Volcano Plot
volcano.ma.plot(diff.res,
sig.value = "pval",
sig.line = 0.05,
plot.type = "volcano",
interactive = F)
# MA Plot
volcano.ma.plot(diff.res,
sig.value = "pval",
sig.line = 0.05,
plot.type = "ma",
interactive = F)


- Merging, resetting, renaming and removing clusters
# let's say you want to merge cluster 3 and 2.
my.obj <- change.clust(my.obj, change.clust = 3, to.clust = 2)
# to reset to the original clusters run this.
my.obj <- change.clust(my.obj, clust.reset = T)
# you can also re-name the cluster numbers to cell types. Remember to reset after this so you can ran other analysis.
my.obj <- change.clust(my.obj, change.clust = 7, to.clust = "B Cell")
# Let's say for what ever reason you want to remove acluster, to do so run this.
my.obj <- clust.rm(my.obj, clust.to.rm = 1)
# Remember that this would perminantly remove the data from all the slots in the object except frrom raw.data slot in the object. If you want to reset you need to start from the filtering cells step in the biginging of the analysis (using cell.filter function).
# To re-position the cells run tSNE again
my.obj <- run.tsne(my.obj, clust.method = "gene.model", gene.list = "my_model_genes.txt")
# Use this for plotting as you make the changes
cluster.plot(my.obj,
cell.size = 1,
plot.type = "tsne",
cell.color = "black",
back.col = "white",
col.by = "clusters",
cell.transparency = 0.5,
clust.dim = 2,
interactive = F)






- Cell gating
my.plot <- gene.plot(my.obj, gene = "GNLY",
plot.type = "scatterplot",
clust.dim = 2,
interactive = F)
cell.gating(my.obj, my.plot = my.plot, plot.type = "tsne")
# or
#my.plot <- cluster.plot(my.obj,
# cell.size = 1,
# cell.transparency = 0.5,
# clust.dim = 2,
# interactive = F)


After downloading the cell ids, use the following command to rename their cluster.
my.obj <- gate.to.clust(my.obj, my.gate = "cellGating.txt", to.clust = 10)1- CPCA (iCellR)** recommended (faster than CCCA)
2- CCCA (iCellR)* recommended
3- MNN (scran wraper) optional
4- MultiCCA (Seurat wraper) optional
5- CPCA + ![]() KNetL based clustering (iCellR)*** recommended for best results!
KNetL based clustering (iCellR)*** recommended for best results!
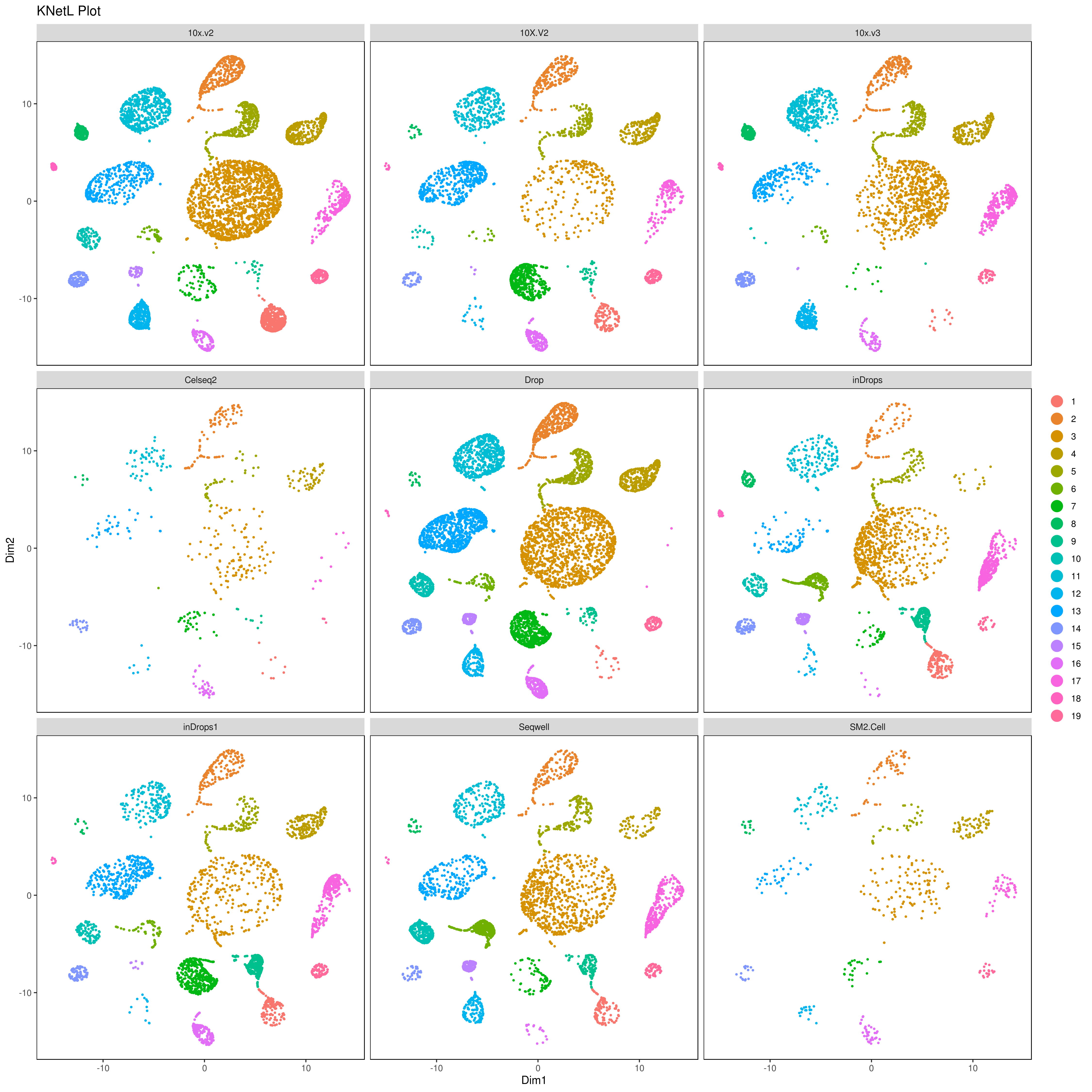
We analyzed nine PBMC sample datasets provided by the Broad Institute to detect batch differences. These datasets were generated using varying technologies, including 10x Chromium v2 (3 samples), 10x Chromium v3, CEL-Seq2, Drop-seq, inDrop, Seq-Well and SMART-Seq. For more info read: https://www.biorxiv.org/content/10.1101/2020.03.31.019109v1.full
## download an object of 9 PBMC samples
sample.file.url = "https://genome.med.nyu.edu/results/external/iCellR/data/pbmc_data/my.obj.Robj"
# download the file
download.file(url = sample.file.url,
destfile = "my.obj.Robj",
method = "auto")
### load iCellR and the object
library(iCellR)
load("my.obj.Robj")
### run PCA on top 2000 genes
my.obj <- run.pca(my.obj, top.rank = 2000)
### find best genes for second round PCA or batch alignment
my.obj <- find.dim.genes(my.obj, dims = 1:30,top.pos = 20, top.neg = 20)
length(my.obj@gene.model)
########### Batch alignment (CPCA method)
my.obj <- iba(my.obj,dims = 1:30, k = 10,ba.method = "CPCA", method = "gene.model", gene.list = my.obj@gene.model)
### impute data
my.obj <- run.impute(my.obj,dims = 1:10,data.type = "pca", nn = 10)
### tSNE and UMAP
my.obj <- run.pc.tsne(my.obj, dims = 1:10)
my.obj <- run.umap(my.obj, dims = 1:10)
### save object
save(my.obj, file = "my.obj.Robj")
### plot
library(gridExtra)
A= cluster.plot(my.obj,plot.type = "umap",interactive = F,cell.size = 0.1)
B= cluster.plot(my.obj,plot.type = "tsne",interactive = F,cell.size = 0.1)
C= cluster.plot(my.obj,plot.type = "umap",col.by = "conditions",interactive = F,cell.size = 0.1)
D=cluster.plot(my.obj,plot.type = "tsne",col.by = "conditions",interactive = F,cell.size = 0.1)
png('AllClusts.png', width = 12, height = 12, units = 'in', res = 300)
grid.arrange(A,B,C,D)
dev.off()
png('AllConds_clusts.png', width = 15, height = 15, units = 'in', res = 300)
cluster.plot(my.obj,
cell.size = 0.5,
plot.type = "umap",
cell.color = "black",
back.col = "white",
cell.transparency = 1,
clust.dim = 2,
interactive = F,cond.facet = T)
dev.off()
genelist = c("PPBP","LYZ","MS4A1","GNLY","FCGR3A","NKG7","CD14","S100A9","CD3E","CD8A","CD4","CD19","IL7R","FOXP3","EPCAM")
for(i in genelist){
MyPlot <- gene.plot(my.obj, gene = i,
interactive = F,
conds.to.plot = NULL,
cell.size = 0.1,
data.type = "main",
plot.data.type = "umap",
scaleValue = T,
min.scale = -2.5,max.scale = 2.0,
cell.transparency = 1)
NameCol=paste("PL",i,sep="_")
eval(call("<-", as.name(NameCol), MyPlot))
}
UMAP = cluster.plot(my.obj,plot.type = "umap",interactive = F,cell.size = 0.1, anno.size=5)
library(cowplot)
filenames <- ls(pattern="PL_")
filenames <- c("UMAP", filenames)
png('genes.png',width = 18, height = 15, units = 'in', res = 300)
plot_grid(plotlist=mget(filenames))
dev.off()



# same as above only change the option to CCCA
my.obj <- iba(my.obj,dims = 1:30, k = 10,ba.method = "CCCA", method = "gene.model", gene.list = my.obj@gene.model)

# same as above only use run.mnn function instead of iba.
###### Run MNN
# This would automatically run all the samples in your experiment
library(scran)
my.obj <- run.mnn(my.obj, k=20, d=50, method = "gene.model", gene.list = my.obj@gene.model)
# detach the scran pacakge after MNN as it masks some of the functions
detach("package:scran", unload=TRUE)

# same as above only use run.anchor function instead of iba.
###### Run Anchor
# This would automatically run all the samples in your experiment
library(Seurat)
my.obj <- run.anchor(my.obj,
normalization.method = "SCT",
scale.factor = 10000,
selection.method = "vst",
nfeatures = 2000,
dims = 1:20)

## download an object of 9 PBMC samples
sample.file.url = "https://genome.med.nyu.edu/results/external/iCellR/example2/my.obj.Robj"
# download the file
download.file(url = sample.file.url,
destfile = "my.obj.Robj",
method = "auto")
### load iCellR and the object
library(iCellR)
load("my.obj.Robj")
### run PCA on top 2000 genes
my.obj <- run.pca(my.obj, top.rank = 2000)
### find best genes for second round PCA or batch alignment
my.obj <- find.dim.genes(my.obj, dims = 1:30,top.pos = 20, top.neg = 20)
length(my.obj@gene.model)
########### Batch alignment (CPCA method)
my.obj <- iba(my.obj,dims = 1:30, k = 10,ba.method = "CPCA", method = "gene.model", gene.list = my.obj@gene.model)
### impute data
my.obj <- run.impute(my.obj,dims = 1:10,data.type = "pca", nn = 10)
### tSNE and UMAP
my.obj <- run.pc.tsne(my.obj, dims = 1:10)
my.obj <- run.umap(my.obj, dims = 1:10)
### run KNetL
my.obj <- run.knetl(my.obj, dims = 1:20, k = 400)
### cluster based on KNetL coordinates
# The object is already clustered but here is an example:
# my.obj <- iclust(my.obj, k = 300, data.type = "knetl")
### save object
save(my.obj, file = "my.obj.Robj")
### plot 1
A= cluster.plot(my.obj,plot.type = "pca",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=F)
B= cluster.plot(my.obj,plot.type = "umap",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=F)
C= cluster.plot(my.obj,plot.type = "tsne",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=F)
D= cluster.plot(my.obj,plot.type = "knetl",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=F)
library(gridExtra)
grid.arrange(A,B,C,D)
### plot 2
cluster.plot(my.obj,
cell.size = 0.5,
plot.type = "knetl",
cell.color = "black",
back.col = "white",
cell.transparency = 1,
clust.dim = 2,
interactive = F,cond.facet = T)
### plot 3
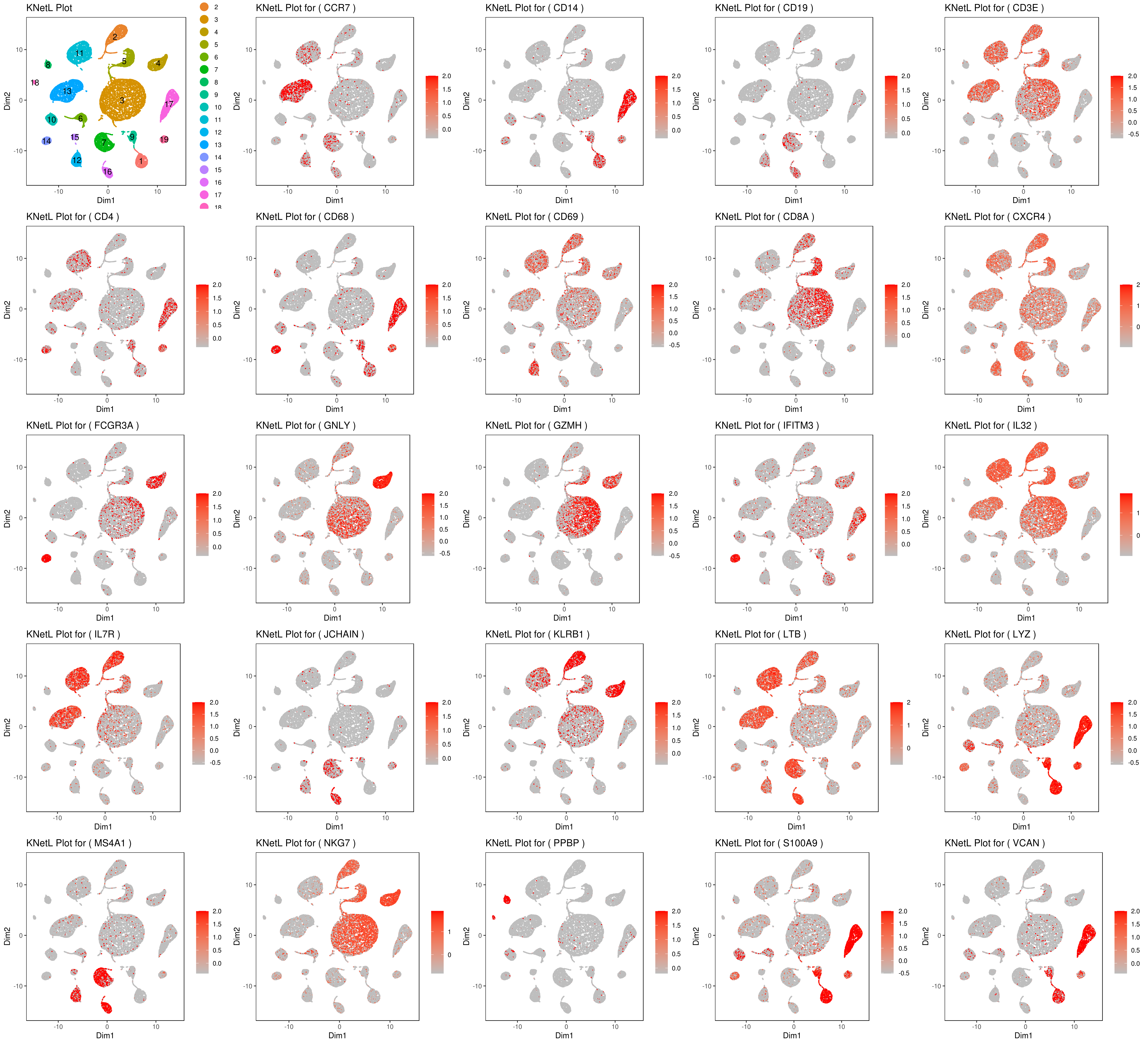
genelist = c("LYZ","MS4A1","GNLY","FCGR3A","NKG7","CD14","S100A9","CD3E","CD8A","CD4","CD19","KLRB1","LTB","IL7R","GZMH","CD68","CCR7","CD68","CD69","CXCR4","IFITM3","IL32","JCHAIN","VCAN","PPBP")
rm(list = ls(pattern="PL_"))
for(i in genelist){
MyPlot <- gene.plot(my.obj, gene = i,
interactive = F,
cell.size = 0.1,
plot.data.type = "knetl",
data.type = "main",
scaleValue = T,
min.scale = -2.5,max.scale = 2.0,
cell.transparency = 1)
NameCol=paste("PL",i,sep="_")
eval(call("<-", as.name(NameCol), MyPlot))
}
library(cowplot)
filenames <- ls(pattern="PL_")
B <- cluster.plot(my.obj,plot.type = "knetl",interactive = F,cell.size = 0.1,cell.transparency = 1,anno.clust=T)
filenames <- c("B",filenames)
plot_grid(plotlist=mget(filenames))
- Pseudotime analysis
MyGenes <- top.markers(marker.genes, topde = 50, min.base.mean = 0.2)
MyGenes <- unique(MyGenes)
pseudotime.tree(my.obj,
marker.genes = MyGenes,
type = "unrooted",
clust.method = "complete")
# or
pseudotime.tree(my.obj,
marker.genes = MyGenes,
type = "classic",
clust.method = "complete")
pseudotime.tree(my.obj,
marker.genes = MyGenes,
type = "jitter",
clust.method = "complete")





- Pseudotime analysis using monocle
library(monocle)
MyMTX <- my.obj@main.data
GeneAnno <- as.data.frame(row.names(MyMTX))
colnames(GeneAnno) <- "gene_short_name"
row.names(GeneAnno) <- GeneAnno$gene_short_name
cell.cluster <- (my.obj@best.clust)
Ha <- data.frame(do.call('rbind', strsplit(as.character(row.names(cell.cluster)),'_',fixed=TRUE)))[1]
clusts <- paste("cl.",as.character(cell.cluster$clusters),sep="")
cell.cluster <- cbind(cell.cluster,Ha,clusts)
colnames(cell.cluster) <- c("Clusts","iCellR.Conds","iCellR.Clusts")
Samp <- new("AnnotatedDataFrame", data = cell.cluster)
Anno <- new("AnnotatedDataFrame", data = GeneAnno)
my.monoc.obj <- newCellDataSet(as.matrix(MyMTX),phenoData = Samp, featureData = Anno)
## find disperesedgenes
my.monoc.obj <- estimateSizeFactors(my.monoc.obj)
my.monoc.obj <- estimateDispersions(my.monoc.obj)
disp_table <- dispersionTable(my.monoc.obj)
unsup_clustering_genes <- subset(disp_table, mean_expression >= 0.1)
my.monoc.obj <- setOrderingFilter(my.monoc.obj, unsup_clustering_genes$gene_id)
# tSNE
my.monoc.obj <- reduceDimension(my.monoc.obj, max_components = 2, num_dim = 10,reduction_method = 'tSNE', verbose = T)
# cluster
my.monoc.obj <- clusterCells(my.monoc.obj, num_clusters = 10)
## plot conditions and clusters based on iCellR analysis
A <- plot_cell_clusters(my.monoc.obj, 1, 2, color = "iCellR.Conds")
B <- plot_cell_clusters(my.monoc.obj, 1, 2, color = "iCellR.Clusts")
## plot clusters based monocle analysis
C <- plot_cell_clusters(my.monoc.obj, 1, 2, color = "Cluster")
# get marker genes from iCellR analysis
MyGenes <- top.markers(marker.genes, topde = 30, min.base.mean = 0.2)
my.monoc.obj <- setOrderingFilter(my.monoc.obj, MyGenes)
my.monoc.obj <- reduceDimension(my.monoc.obj, max_components = 2,method = 'DDRTree')
# order cells
my.monoc.obj <- orderCells(my.monoc.obj)
# plot based on iCellR analysis and marker genes from iCellR
D <- plot_cell_trajectory(my.monoc.obj, color_by = "iCellR.Clusts")
## heatmap genes from iCellR
plot_pseudotime_heatmap(my.monoc.obj[MyGenes,],
cores = 1,
cluster_rows = F,
use_gene_short_name = T,
show_rownames = T)


# Read an example file
my.hto <- read.table(file = system.file('extdata', 'dense_umis.tsv', package = 'iCellR'), as.is = TRUE)
# see the head of the file for the first few columns
head(my.hto)[1:3]
# TGACAACAGGGCTCTC AAGGAGCGTCATTAGC AGTGAGGAGACTGTAA
#Hashtag1-GTCAACTCTTTAGCG 3 7 7
#Hashtag2-TGATGGCCTATTGGG 18 24 1761
#Hashtag3-TTCCGCCTCTCTTTG 7 8 5
#Hashtag4-AGTAAGTTCAGCGTA 0 0 0
#Hashtag5-AAGTATCGTTTCGCA 890 2 11
#Hashtag7-TGTCTTTCCTGCCAG 5 3 3
# run annotation
htos <- hto.anno(hto.data = my.hto, cov.thr = 10, assignment.thr = 80)
head(htos)
# Hashtag1-GTCAACTCTTTAGCG Hashtag2-TGATGGCCTATTGGG
#TGACAACAGGGCTCTC 3 18
#AAGGAGCGTCATTAGC 7 24
#AGTGAGGAGACTGTAA 7 1761
#ATCCACCCATGTTCCC 753 20
#AAACGGGCAGGACCCT 728 24
#ATGTGTGAGTCTTGCA 4 25
# Hashtag3-TTCCGCCTCTCTTTG Hashtag4-AGTAAGTTCAGCGTA
#TGACAACAGGGCTCTC 7 0
#AAGGAGCGTCATTAGC 8 0
#AGTGAGGAGACTGTAA 5 0
#ATCCACCCATGTTCCC 3 0
#AAACGGGCAGGACCCT 3 0
#ATGTGTGAGTCTTGCA 370 0
# Hashtag5-AAGTATCGTTTCGCA Hashtag7-TGTCTTTCCTGCCAG unmapped
#TGACAACAGGGCTCTC 890 5 17
#AAGGAGCGTCATTAGC 2 3 3
#AGTGAGGAGACTGTAA 11 3 87
#ATCCACCCATGTTCCC 5 6 18
#AAACGGGCAGGACCCT 9 3 16
#ATGTGTGAGTCTTGCA 9 1011 25
# assignment.annotation percent.match coverage low.cov
#TGACAACAGGGCTCTC Hashtag5-AAGTATCGTTTCGCA 94.68085 940 FALSE
#AAGGAGCGTCATTAGC Hashtag2-TGATGGCCTATTGGG 51.06383 47 TRUE
#AGTGAGGAGACTGTAA Hashtag2-TGATGGCCTATTGGG 93.97012 1874 FALSE
#ATCCACCCATGTTCCC Hashtag1-GTCAACTCTTTAGCG 93.54037 805 FALSE
#AAACGGGCAGGACCCT Hashtag1-GTCAACTCTTTAGCG 92.97573 783 FALSE
#ATGTGTGAGTCTTGCA Hashtag7-TGTCTTTCCTGCCAG 70.01385 1444 FALSE
# assignment.threshold
#TGACAACAGGGCTCTC good.assignment
#AAGGAGCGTCATTAGC unsure
#AGTGAGGAGACTGTAA good.assignment
#ATCCACCCATGTTCCC good.assignment
#AAACGGGCAGGACCCT good.assignment
#ATGTGTGAGTCTTGCA unsure
# plot
A = ggplot(htos, aes(assignment.annotation,percent.match)) +
geom_jitter(alpha = 0.25, color = "blue") +
geom_boxplot(alpha = 0.5) +
theme_bw() +
theme(axis.text.x=element_text(angle=90))
B = ggplot(htos, aes(low.cov,percent.match)) +
geom_jitter(alpha = 0.25, color = "blue") +
geom_boxplot(alpha = 0.5) +
theme_bw() +
theme(axis.text.x=element_text(angle=90))
library(gridExtra)
png('HTOs.png', width = 8, height = 8, units = 'in', res = 300)
grid.arrange(A,B,ncol=2)
dev.off()

- Filtering HTOs and merging the samples
# let's say you decided filtering based on 80%
dim(htos)
# [1] 1500 12
htos <- subset(htos,htos$percent.match > 80)
dim(htos)
# [1] 1073 12
# Take the cell IDs from Hashtag1
sample1 <- row.names(subset(htos,htos$assignment.annotation == "Hashtag1-GTCAACTCTTTAGCG"))
head(sample1)
# [1] "ATCCACCCATGTTCCC" "AAACGGGCAGGACCCT" "TTCTACATCCTCATTA" "GGTATTGTCCTATGTT"
# [5] "GTCGTAATCTTACCTA" "ACAGCCGGTTGGGACA"
length(sample1)
# [1] 213
# in this case you have 213 cells in sample 1 (Hashtag1)
# Take the cell IDs from Hashtag2
sample2 <- row.names(subset(htos,htos$assignment.annotation == "Hashtag2-TGATGGCCTATTGGG"))
# now read your RNA data
# example:
RNA.data <- load10x("YOUR/data/filtered_gene_bc_matrices/hg19/")
head(RNA.data)[1:2]
# AAACATAAAACCAG CCCCATTGAGCTAA
#A1BG.AS1 0 0
#BCLA 0 0
#A2M 0 0
#GATA1 0 0
# NOTE: the RNA data has the cell IDs in the same format as HTOs
# "AAACATAAAACCAG" "CCCCATTGAGCTAA" ...
# Not "AAACATAAAACCAG.1" "CCCCATTGAGCTAA.1" ...
# demultiplex RNA data
# Take RNA-Seq data with the cell IDs from Hashtag1
sample1.rna <- RNA.data[ , which(names(RNA.data) %in% sample1)]
# Take RNA-Seq data with the cell IDs from Hashtag2
sample2.rna <- RNA.data[ , which(names(RNA.data) %in% sample2)]
# aggregate (merge the 2 or more samples after demultiplexing)
my.data <- data.aggregation(samples = c("sample1.rna","sample2.rna"),
condition.names = c("S1","S2"))
# make iCellR object
my.obj <- make.obj(my.data)
# The rest is as above :)This data is from this publication (GEO number: GSE156246 and PMID: 34911733)
This is a how to guide to run i.score function in iCellR and to reproduce the above published data for G0 and non G0 cells.
Download the sample iCellR objects (used in the publication) from here: https://genome.med.nyu.edu/results/external/iCellR/i.score/ ([email protected] in these objects are log normalized)
Download sample gene signatures from here: https://genome.med.nyu.edu/results/external/iCellR/i.score/gene_signatures.tar.gz (gene signatures used in the publication are in the supplementary data of the paper)
# load sample gene signature that are in iCellR
# (these cell cycle signatures are from here: https://www.nature.com/articles/s41586-019-1884-x)
library(iCellR)
G0 <- readLines(system.file('extdata', 'G0.txt', package = 'iCellR'))
G1S <- readLines(system.file('extdata', 'G1S.txt', package = 'iCellR'))
G2M <- readLines(system.file('extdata', 'G2M.txt', package = 'iCellR'))
M <- readLines(system.file('extdata', 'M.txt', package = 'iCellR'))
MG1 <- readLines(system.file('extdata', 'MG1.txt', package = 'iCellR'))
S <- readLines(system.file('extdata', 'S.txt', package = 'iCellR'))
# load all the gene signatures
Melnick_10_GILMORE_CORE_NFKB_PATHWAY.txt <- readLines("10_GILMORE_CORE_NFKB_PATHWAY.txt")
Melnick_11_HALLMARK_MYC_TARGETS_V1.txt <- readLines("11_HALLMARK_MYC_TARGETS_V1.txt")
Melnick_12_GO_BETA_CATENIN_BINDING.txt <- readLines("12_GO_BETA_CATENIN_BINDING.txt")
Melnick_13_PID_BETA_CATENIN_NUC_PATHWAY.txt <- readLines("13_PID_BETA_CATENIN_NUC_PATHWAY.txt")
Melnick_14_PID_WNT_SIGNALING_PATHWAY.txt <- readLines("14_PID_WNT_SIGNALING_PATHWAY.txt")
Melnick_15_PID_WNT_CANONICAL_PATHWAY.txt <- readLines("15_PID_WNT_CANONICAL_PATHWAY.txt")
Melnick_16_Pribluda_SENESCENCE_INFLAMMATORY_GENES.txt <- readLines("16_Pribluda_SENESCENCE_INFLAMMATORY_GENES.txt")
Melnick_17_FRIDMAN_SENESCENCE_DN.txt <- readLines("17_FRIDMAN_SENESCENCE_DN.txt")
Melnick_18_FRIDMAN_SENESCENCE_UP.txt <- readLines("18_FRIDMAN_SENESCENCE_UP.txt")
Melnick_19_DeJONGE_LSC_TOP50_genes.txt <- readLines("19_DeJONGE_LSC_TOP50_genes.txt")
Melnick_1_AML1566_AraC_UP.txt <- readLines("1_AML1566_AraC_UP.txt")
Melnick_20_GAL_LEUKEMIC_STEM_CELL_UP.txt <- readLines("20_GAL_LEUKEMIC_STEM_CELL_UP.txt")
Melnick_21_GAL_LEUKEMIC_STEM_CELL_DN.txt <- readLines("21_GAL_LEUKEMIC_STEM_CELL_DN.txt")
Melnick_22_EPPERT_CE_HSC_LSC.txt <- readLines("22_EPPERT_CE_HSC_LSC.txt")
Melnick_23_JAATINEN_HEMATOPOIETIC_STEM_CELL_UP.txt <- readLines("23_JAATINEN_HEMATOPOIETIC_STEM_CELL_UP.txt")
Melnick_24_JAATINEN_HEMATOPOIETIC_STEM_CELL_DN.txt <- readLines("24_JAATINEN_HEMATOPOIETIC_STEM_CELL_DN.txt")
Melnick_25_INFLAMMATORY_RESPONSE.txt <- readLines("25_INFLAMMATORY_RESPONSE.txt")
Melnick_26_RAMALHO_STEMNESS_DN.txt <- readLines("26_RAMALHO_STEMNESS_DN.txt")
Melnick_27_RAMALHO_STEMNESS_UP.txt <- readLines("27_RAMALHO_STEMNESS_UP.txt")
Melnick_28_REACTOME_REGULATION_OF_MITOTIC_CELL_CYCLE.txt <- readLines("28_REACTOME_REGULATION_OF_MITOTIC_CELL_CYCLE.txt")
Melnick_2_AML1566_AraC_DN.txt <- readLines("2_AML1566_AraC_DN.txt")
Melnick_3_DUY_CISG_UP.txt <- readLines("3_DUY_CISG_UP.txt")
Melnick_4_DUY_CISG_DN.txt <- readLines("4_DUY_CISG_DN.txt")
Melnick_5_DIAPAUSE_UP_BOROVIAK.txt <- readLines("5_DIAPAUSE_UP_BOROVIAK.txt")
Melnick_6_BOROVIAK_DIAPAUSE_DN.txt <- readLines("6_BOROVIAK_DIAPAUSE_DN.txt")
Melnick_7_SASP_COPPE.txt <- readLines("7_SASP_COPPE.txt")
Melnick_8_SALDIVAR_ATR_SUPPRESSED_TARGETS.txt <- readLines("8_SALDIVAR_ATR_SUPPRESSED_TARGETS.txt")
Melnick_9_BIOCARTA_NFKB_PATHWAY.txt <- readLines("9_BIOCARTA_NFKB_PATHWAY.txt")
diapause_neg.txt <- readLines("diapause_neg.txt")
diapause_pos_and_neg.txt <- readLines("diapause_pos_and_neg.txt")
diapause_pos.txt <- readLines("diapause_pos.txt")
DTP_sig_150_Down.txt <- readLines("DTP_sig_150_Down.txt")
DTP_sig_150_up.txt <- readLines("DTP_sig_150_up.txt")
Lum_uniq_down.txt <- readLines("Lum_uniq_down.txt")
Lum_uniq_up.txt <- readLines("Lum_uniq_up.txt")
Mes_uniq_down.txt <- readLines("Mes_uniq_down.txt")
Mes_uniq_up.txt <- readLines("Mes_uniq_up.txt")
panDTP_DN.txt <- readLines("new_panDTP_DN.txt")
panDTP_up.txt <- readLines("new_panDTP_up.txt")
mes_DTP_included_DEG_DN.txt <- readLines("new_mes_DTP_included_DEG_DN.txt")
mes_DTP_included_DEG_UP.txt <- readLines("new_mes_DTP_included_DEG_UP.txt")
lum_DTP_included_DEG_DN.txt <- readLines("new_lum_DTP_included_DEG_DN.txt")
lum_DTP_included_DEG_UP.txt <- readLines("new_lum_DTP_included_DEG_UP.txt")
lum_DTP_specific_UP_noCC.txt <- readLines("new_lum_DTP_specific_UP_noCC_.txt")
mes_DTP_specific_UP_noCC.txt <- readLines("new_mes_DTP_specific_UP_noCC_.txt")Group all the signatures in one character object:
All <- c("Melnick_10_GILMORE_CORE_NFKB_PATHWAY.txt","Melnick_11_HALLMARK_MYC_TARGETS_V1.txt","Melnick_12_GO_BETA_CATENIN_BINDING.txt","Melnick_13_PID_BETA_CATENIN_NUC_PATHWAY.txt","Melnick_14_PID_WNT_SIGNALING_PATHWAY.txt","Melnick_15_PID_WNT_CANONICAL_PATHWAY.txt","Melnick_16_Pribluda_SENESCENCE_INFLAMMATORY_GENES.txt","Melnick_17_FRIDMAN_SENESCENCE_DN.txt","Melnick_18_FRIDMAN_SENESCENCE_UP.txt","Melnick_19_DeJONGE_LSC_TOP50_genes.txt","Melnick_1_AML1566_AraC_UP.txt","Melnick_20_GAL_LEUKEMIC_STEM_CELL_UP.txt","Melnick_21_GAL_LEUKEMIC_STEM_CELL_DN.txt","Melnick_22_EPPERT_CE_HSC_LSC.txt","Melnick_23_JAATINEN_HEMATOPOIETIC_STEM_CELL_UP.txt","Melnick_24_JAATINEN_HEMATOPOIETIC_STEM_CELL_DN.txt","Melnick_25_INFLAMMATORY_RESPONSE.txt","Melnick_26_RAMALHO_STEMNESS_DN.txt","Melnick_27_RAMALHO_STEMNESS_UP.txt","Melnick_28_REACTOME_REGULATION_OF_MITOTIC_CELL_CYCLE.txt","Melnick_2_AML1566_AraC_DN.txt","Melnick_3_DUY_CISG_UP.txt","Melnick_4_DUY_CISG_DN.txt","Melnick_5_DIAPAUSE_UP_BOROVIAK.txt","Melnick_6_BOROVIAK_DIAPAUSE_DN.txt","Melnick_7_SASP_COPPE.txt","Melnick_8_SALDIVAR_ATR_SUPPRESSED_TARGETS.txt","Melnick_9_BIOCARTA_NFKB_PATHWAY.txt","diapause_neg.txt","diapause_pos_and_neg.txt","diapause_pos.txt","DTP_sig_150_Down.txt","DTP_sig_150_up.txt","Lum_uniq_down.txt","Lum_uniq_up.txt","Mes_uniq_down.txt","Mes_uniq_up.txt","G0","G1S","G2M","M","MG1","S","panDTP_DN.txt","panDTP_up.txt","mes_DTP_included_DEG_DN.txt","mes_DTP_included_DEG_UP.txt","lum_DTP_included_DEG_DN.txt","lum_DTP_included_DEG_UP.txt","lum_DTP_specific_UP_noCC.txt","mes_DTP_specific_UP_noCC.txt")Load your sample iCellR object
load("BT474_DTP.Robj")Score for cell cycle gene signatures with any of the following scoring methods: tirosh, mean, sum, gsva, ssgsea, zscore and plage. (tirosh and zscore methods are recommended to perform best)
dat1 <- i.score(my.obj, scoring.List = c("G0","G1S","G2M","M","MG1","S") ,scoring.method = "tirosh",return.stats = TRUE, data.type = "raw.data")
write.table(dat1,"tirosh_G0.tsv",sep="\t")Score for all the other signatures (tirosh, mean, sum, gsva, ssgsea, zscore and plage)
dat2 <- i.score(my.obj, scoring.List = All ,scoring.method = "tirosh",return.stats = TRUE, data.type = "raw.data")
write.table(dat2,"tirosh_all.tsv",sep="\t")Prepare data to plot (marge dat1 and dat2)
dir.create("boxplots_tirosh")
setwd("boxplots_tirosh")
data <- read.table("../tirosh_all.tsv",sep="\t",header=T)
dataCC <- read.table("../tirosh_G0.tsv",sep="\t",header=T)
df = as.character(dataCC$assignment.annotation) == "G0"
df[ df == "TRUE" ] <- "GO"
df[ df == "FALSE" ] <- "nonGO"
data <- cbind(cond = rep("sample",length(df)),
ID = rownames(data),
assignment.annotation = dataCC$assignment.annotation,
GO_nonGO = df,
data)
write.table((data),file="data.xls",sep="\t", row.names =F)Plot all the signatures individually:
data <- read.table("data.xls",sep="\t",header=T)
g <- head(data)[5:55]
g <- colnames(g)
library(ggpubr)
for(i in g){
name <- paste("boxplot_",i,".png",sep="")
png(name,width = 6, height = 4, units = 'in', res = 300)
print(ggplot(data, aes(x= GO_nonGO,y=data[, i],fill = GO_nonGO, alpha = 0.5)) +
geom_jitter(size = 0.2, color="black") +
geom_violin(trim=FALSE, col = "black", alpha = 0.5) +
geom_boxplot(outlier.color = NA) +
theme_bw() +
xlab("Condition") +
ylab("Signature Score") +
scale_y_continuous(trans = "log1p") +
stat_compare_means(aes(group = GO_nonGO), label = "p.signif", label.x = 1.5) +
theme(axis.text.x = element_blank()))
dev.off()
}Example for "lum_DTP_included_DEG_DN.txt"
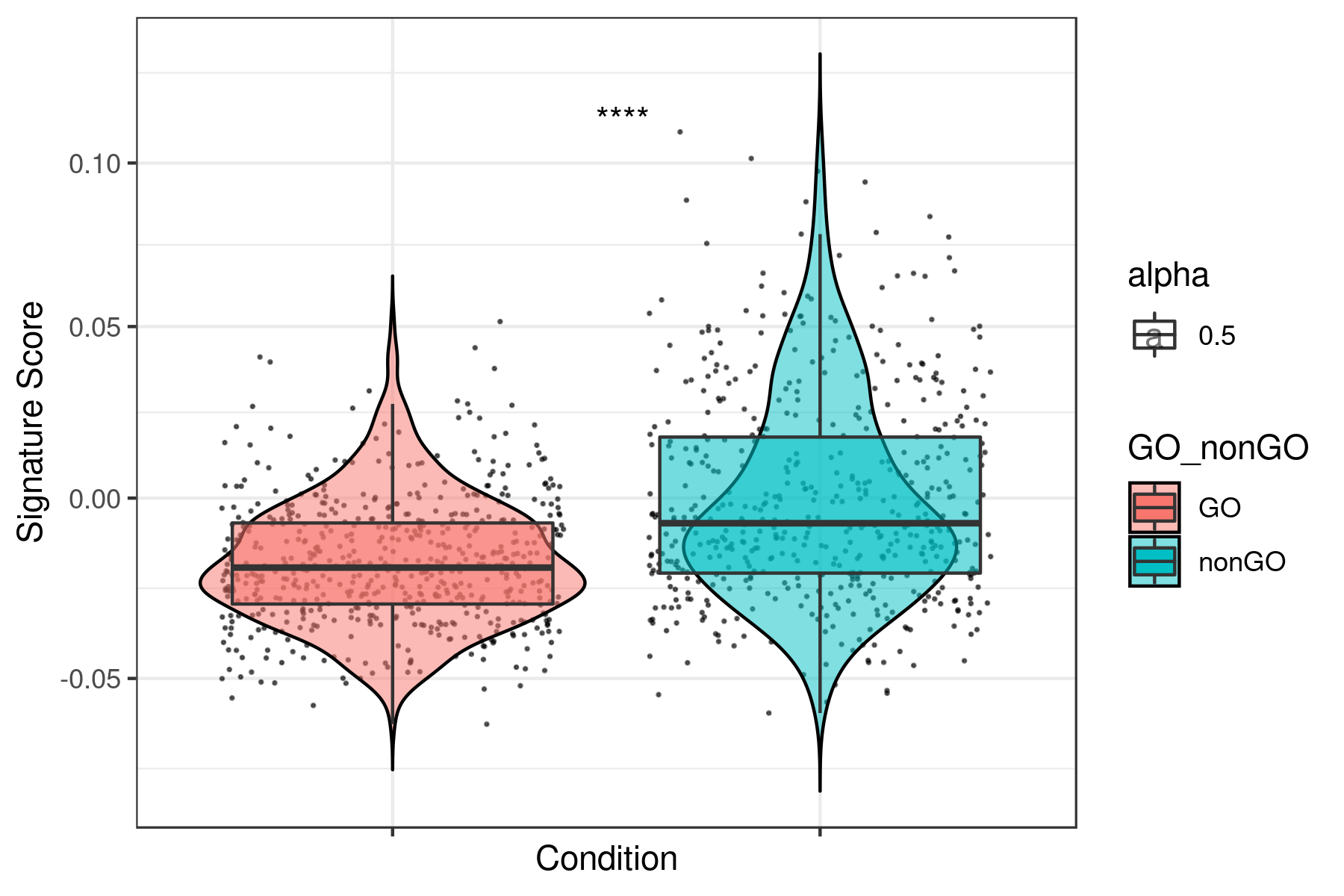
To see all the plots made as above go to this link: https://genome.med.nyu.edu/results/external/iCellR/i.score/test/boxplots_tirosh/
- Download test samples
sample.file.url = "https://genome.med.nyu.edu/results/external/iCellR/data/CITE-Seq_sample_RNA.tsv.gz"
# download RNA file
download.file(url = sample.file.url,
destfile = "CITE-Seq_sample_RNA.tsv.gz",
method = "auto")
sample.file.url = "https://genome.med.nyu.edu/results/external/iCellR/data/CITE-Seq_sample_ADT.tsv.gz"
# download ADT file
download.file(url = sample.file.url,
destfile = "CITE-Seq_sample_ADT.tsv.gz",
method = "auto") - Read the files and make your object
# Read RNA file
rna.data <- read.delim("CITE-Seq_sample_RNA.tsv.gz",header=TRUE)
# see the head
head(rna.data)[1:3]
# CTGTTTACACCGCTAG CTCTACGGTGTGGCTC AGCAGCCAGGCTCATT
#A1BG 0 0 0
#A1BG-AS1 0 0 0
#A1CF 0 0 0
#A2M 0 0 0
#A2M-AS1 0 0 0
#A2ML1 0 0 0
# Read ADT file
adt.data <- read.delim("CITE-Seq_sample_ADT.tsv.gz",header=TRUE)
# see the head
head(adt.data)[1:3]
# CTGTTTACACCGCTAG CTCTACGGTGTGGCTC AGCAGCCAGGCTCATT
#CD3 60 52 89
#CD4 72 49 112
#CD8 76 59 61
#CD45RA 575 3943 682
#CD56 64 68 87
#CD16 161 107 117
# if you had multiple sample use the data.aggregation function for both RNA and ADT data.
# make iCellR object
my.obj <- make.obj(rna.data)
# check object
my.obj
###################################
,--. ,-----. ,--.,--.,------.
`--'' .--./ ,---. | || || .--. '
,--.| | | .-. :| || || '--'.'
| |' '--'\ --. | || || |
`--' `-----' `----'`--'`--'`--' '--'
###################################
An object of class iCellR version: 1.1.4
Raw/original data dimentions (rows,columns): 20501,8617
Data conditions: no conditions/single sample
Row names: A1BG,A1BG-AS1,A1CF ...
Columns names: CTGTTTACACCGCTAG,CTCTACGGTGTGGCTC,AGCAGCCAGGCTCATT ...
###################################
QC stats performed:FALSE, PCA performed:FALSE, CCA performed:FALSE
Clustering performed:FALSE, Number of clusters:0
tSNE performed:FALSE, UMAP performed:FALSE, DiffMap performed:FALSE
Main data dimentions (rows,columns):0,0
Normalization factors:,...
Imputed data dimentions (rows,columns):0,0
############## scVDJ-Seq ###########
VDJ data dimentions (rows,columns):0,0
############## CITE-Seq ############
ADT raw data dimentions (rows,columns):0,0
ADT main data dimentions (rows,columns):0,0
ADT columns names:...
ADT row names:...
########### iCellR object ##########- add ADT data
my.obj <- add.adt(my.obj, adt.data = adt.data)
# check too see
my.obj
###################################
,--. ,-----. ,--.,--.,------.
`--'' .--./ ,---. | || || .--. '
,--.| | | .-. :| || || '--'.'
| |' '--'\ --. | || || |
`--' `-----' `----'`--'`--'`--' '--'
###################################
An object of class iCellR version: 1.1.4
Raw/original data dimentions (rows,columns): 20501,8617
Data conditions: no conditions/single sample
Row names: A1BG,A1BG-AS1,A1CF ...
Columns names: CTGTTTACACCGCTAG,CTCTACGGTGTGGCTC,AGCAGCCAGGCTCATT ...
###################################
QC stats performed:FALSE, PCA performed:FALSE, CCA performed:FALSE
Clustering performed:FALSE, Number of clusters:0
tSNE performed:FALSE, UMAP performed:FALSE, DiffMap performed:FALSE
Main data dimentions (rows,columns):0,0
Normalization factors:,...
Imputed data dimentions (rows,columns):0,0
############## scVDJ-Seq ###########
VDJ data dimentions (rows,columns):0,0
############## CITE-Seq ############
- ADT raw data dimentions (rows,columns):10,8617
ADT main data dimentions (rows,columns):0,0
ADT columns names:...
ADT row names:...
########### iCellR object ##########- QC, filter, normalize, merge ADT and RNA data, run PCA and UMAP
# QC
my.obj <- qc.stats(my.obj,
s.phase.genes = s.phase,
g2m.phase.genes = g2m.phase)
# plot as mentioned above
# filter
my.obj <- cell.filter(my.obj,
min.mito = 0,
max.mito = 0.07 ,
min.genes = 500,
max.genes = 4000,
min.umis = 0,
max.umis = Inf)
# normalize RNA
my.obj <- norm.data(my.obj, norm.method = "ranked.glsf", top.rank = 500)
# normalize ADT
my.obj <- norm.adt(my.obj)
# gene stats
my.obj <- gene.stats(my.obj, which.data = "main.data")
# find genes for PCA
my.obj <- make.gene.model(my.obj, my.out.put = "data",
dispersion.limit = 1.5,
base.mean.rank = 500,
no.mito.model = T,
mark.mito = T,
interactive = F,
no.cell.cycle = T,
out.name = "gene.model")
# merge RNA and ADT data
my.obj <- adt.rna.merge(my.obj, adt.data = "main")
# run PCA and the rest is as above
my.obj <- run.pca(my.obj, method = "gene.model", gene.list = my.obj@gene.model,data.type = "main")
# 2 pass PCA
my.obj <- find.dim.genes(my.obj, dims = 1:20,top.pos = 20, top.neg = 20)
# second round PC
my.obj <- run.pca(my.obj, method = "gene.model", gene.list = my.obj@gene.model,data.type = "main")
my.obj <- run.umap(my.obj, dims = 1:10)
# check your object
my.obj
###################################
,--. ,-----. ,--.,--.,------.
`--'' .--./ ,---. | || || .--. '
,--.| | | .-. :| || || '--'.'
| |' '--'\ --. | || || |
`--' `-----' `----'`--'`--'`--' '--'
###################################
An object of class iCellR version: 1.1.4
Raw/original data dimentions (rows,columns): 20501,8617
Data conditions: no conditions/single sample
Row names: A1BG,A1BG-AS1,A1CF ...
Columns names: CTGTTTACACCGCTAG,CTCTACGGTGTGGCTC,AGCAGCCAGGCTCATT ...
###################################
QC stats performed:TRUE, PCA performed:TRUE, CCA performed:FALSE
Clustering performed:TRUE, Number of clusters:14
tSNE performed:FALSE, UMAP performed:TRUE, DiffMap performed:FALSE
Main data dimentions (rows,columns):20511,8305
Normalization factors:8.448547776071,...
Imputed data dimentions (rows,columns):0,0
############## scVDJ-Seq ###########
VDJ data dimentions (rows,columns):0,0
############## CITE-Seq ############
ADT raw data dimentions (rows,columns):10,8617
ADT main data dimentions (rows,columns):10,8617
ADT columns names:CTGTTTACACCGCTAG...
ADT row names:ADT_CD3...
########### iCellR object ##########- plot
# find ADT gene names
grep("^ADT_", rownames(my.obj@main.data),value=T)
# [1] "ADT_CD3" "ADT_CD4" "ADT_CD8" "ADT_CD45RA" "ADT_CD56"
# [6] "ADT_CD16" "ADT_CD11c" "ADT_CD14" "ADT_CD19" "ADT_CD34"
A = gene.plot(my.obj,
gene = "ADT_CD3",
plot.data.type = "umap",
interactive = F,
cell.transparency = 0.5)
B = gene.plot(my.obj,
gene = "CD3E",
plot.data.type = "umap",
interactive = F,
cell.transparency = 0.5)
C = gene.plot(my.obj,
gene = "ADT_CD16",
plot.data.type = "umap",
interactive = F,
cell.transparency = 0.5)
D = gene.plot(my.obj,
gene = "FCGR3A",
plot.data.type = "umap",
interactive = F,
cell.transparency = 0.5)
library(gridExtra)
grid.arrange(A,B,C,D)

Here is an example of how to add VDJ data.
###### an example file
my.vdj <- read.csv(file = system.file('extdata', 'all_contig_annotations.csv',
package = 'iCellR'),
as.is = TRUE)
###
head(my.vdj)
# barcode is_cell contig_id high_confidence length
#1 AAACCTGTCCGAACGC-1 True AAACCTGTCCGAACGC-1_contig_1 True 654
#2 AAACCTGTCCGAACGC-1 True AAACCTGTCCGAACGC-1_contig_2 True 697
#3 AAACCTGTCCGAACGC-1 True AAACCTGTCCGAACGC-1_contig_3 False 496
#4 AAACCTGTCCGAACGC-1 True AAACCTGTCCGAACGC-1_contig_4 True 539
#5 AAACCTGTCGATGAGG-1 True AAACCTGTCGATGAGG-1_contig_1 True 705
#6 AAACCTGTCGATGAGG-1 True AAACCTGTCGATGAGG-1_contig_2 True 491
# chain v_gene d_gene j_gene c_gene full_length productive cdr3
#1 TRB TRBV4-1 None TRBJ2-7 TRBC2 True True CASSQGVEQYF
#2 TRA TRAV8-1 None TRAJ42 TRAC True True CAVKGGSQGNLIF
#3 TRB None None TRBJ1-4 TRBC1 False None None
#4 Multi None None TRAJ10 TRBC1 False None None
#5 TRB TRBV5-5 TRBD1 TRBJ2-7 TRBC1 True True CASSLVSGGNEQYF
#6 TRB None None TRBJ1-2 TRBC1 False None None
# cdr3_nt reads umis raw_clonotype_id
#1 TGCGCCAGCAGCCAAGGGGTCGAGCAGTACTTC 42610 19 clonotype150
#2 TGTGCCGTGAAGGGAGGAAGCCAAGGAAATCTCATCTTT 12297 4 clonotype150
#3 None 4314 1 clonotype150
#4 None 2212 1 clonotype150
#5 TGTGCCAGCAGCTTGGTCTCAGGGGGAAACGAGCAGTACTTC 21148 8 clonotype2
#6 None 17717 16 clonotype2
# raw_consensus_id
#1 clonotype150_consensus_1
#2 clonotype150_consensus_2
#3 None
#4 None
#5 clonotype2_consensus_1
#6 None
#### Prepare the vdj file
My.VDJ <- prep.vdj(vdj.data = my.vdj, cond.name = "NULL")
###
head(My.VDJ)
# raw_clonotype_id barcode is_cell contig_id
#1 clonotype1 ACGCCAGCAAGCGCTC.1 True ACGCCAGCAAGCGCTC-1_contig_2
#2 clonotype1 AACGTTGAGTACGATA.1 True AACGTTGAGTACGATA-1_contig_2
#3 clonotype1 AACTCTTGTCAAAGCG.1 True AACTCTTGTCAAAGCG-1_contig_1
#4 clonotype1 AACGTTGAGTACGATA.1 True AACGTTGAGTACGATA-1_contig_1
#5 clonotype1 ACGCCAGCAAGCGCTC.1 True ACGCCAGCAAGCGCTC-1_contig_1
#6 clonotype1 ACGATGTTCTGGTATG.1 True ACGATGTTCTGGTATG-1_contig_2
# high_confidence length chain v_gene d_gene j_gene c_gene full_length
#1 True 571 TRA TRAV27 None TRAJ37 TRAC True
#2 True 730 TRA TRAV27 None TRAJ37 TRAC True
#3 True 722 TRB TRBV6-3 TRBD2 TRBJ1-1 TRBC1 True
#4 True 723 TRB TRBV6-3 TRBD2 TRBJ1-1 TRBC1 True
#5 True 722 TRB TRBV6-3 TRBD2 TRBJ1-1 TRBC1 True
#6 True 726 TRA TRAV27 None TRAJ37 TRAC True
# productive cdr3 cdr3_nt reads
#1 True CAGGRSSNTGKLIF TGTGCAGGAGGACGCTCTAGCAACACAGGCAAACTAATCTTT 14241
#2 True CAGGRSSNTGKLIF TGTGCAGGAGGACGCTCTAGCAACACAGGCAAACTAATCTTT 27679
#3 True CASRTGAGATEAFF TGTGCCAGCAGGACCGGGGCGGGAGCCACTGAAGCTTTCTTT 51844
#4 True CASRTGAGATEAFF TGTGCCAGCAGGACCGGGGCGGGAGCCACTGAAGCTTTCTTT 38120
#5 True CASRTGAGATEAFF TGTGCCAGCAGGACCGGGGCGGGAGCCACTGAAGCTTTCTTT 24635
#6 True CAGGRSSNTGKLIF TGTGCAGGAGGACGCTCTAGCAACACAGGCAAACTAATCTTT 13720
# umis raw_consensus_id my.raw_clonotype_id clonotype.Freq proportion
#1 8 clonotype1_consensus_2 clonotype1 43 0.1572212
#2 10 clonotype1_consensus_2 clonotype1 43 0.1572212
#3 24 clonotype1_consensus_1 clonotype1 43 0.1572212
#4 23 clonotype1_consensus_1 clonotype1 43 0.1572212
#5 11 clonotype1_consensus_1 clonotype1 43 0.1572212
#6 7 clonotype1_consensus_2 clonotype1 43 0.1572212
# total.colonotype
#1 109
#2 109
#3 109
#4 109
#5 109
#6 109
####
png('vdj.stats.png',width = 16, height = 8, units = 'in', res = 300)
vdj.stats(My.VDJ)
dev.off()
### add vdj data to you object
my.obj <- add.vdj(demo.obj, vdj.data = My.VDJ)

Another example with multiple files
# first prepare the files.
# this function would filter the files, calculate clonotype frequencies and proportions and add conditions to the cell ids.
my.vdj.1 <- prep.vdj(vdj.data = "all_contig_annotations.csv", cond.name = "WT")
my.vdj.2 <- prep.vdj(vdj.data = "all_contig_annotations.csv", cond.name = "KO")
my.vdj.3 <- prep.vdj(vdj.data = "all_contig_annotations.csv", cond.name = "Ctrl")
# concatenate all the conditions
my.vdj.data <- rbind(my.vdj.1, my.vdj.2, my.vdj.3)
# see head of the file
head(my.vdj.data)
# raw_clonotype_id barcode is_cell contig_id
#1 clonotype1 WT_AAACCTGAGCTAACTC-1 True AAACCTGAGCTAACTC-1_contig_1
#2 clonotype1 WT_AAACCTGAGCTAACTC-1 True AAACCTGAGCTAACTC-1_contig_2
#3 clonotype1 WT_AGTTGGTTCTCGCATC-1 True AGTTGGTTCTCGCATC-1_contig_3
#4 clonotype1 WT_TGACAACCAACTGCTA-1 True TGACAACCAACTGCTA-1_contig_1
#5 clonotype1 WT_TGTCCCAGTCAAACTC-1 True TGTCCCAGTCAAACTC-1_contig_1
#6 clonotype1 WT_TGTCCCAGTCAAACTC-1 True TGTCCCAGTCAAACTC-1_contig_2
# high_confidence length chain v_gene d_gene j_gene c_gene full_length
#1 True 693 TRA TRAV8-1 None TRAJ21 TRAC True
#2 True 744 TRB TRBV28 TRBD1 TRBJ2-1 TRBC2 True
#3 True 647 TRA TRAV8-1 None TRAJ21 TRAC True
#4 True 508 TRB TRBV28 TRBD1 TRBJ2-1 TRBC2 True
#5 True 660 TRA TRAV8-1 None TRAJ21 TRAC True
#6 True 770 TRB TRBV28 TRBD1 TRBJ2-1 TRBC2 True
# productive cdr3 cdr3_nt
#1 True CAVKDFNKFYF TGTGCCGTGAAAGACTTCAACAAATTTTACTTT
#2 True CASSLFSGTGTNEQFF TGTGCCAGCAGTTTATTTTCCGGGACAGGGACGAATGAGCAGTTCTTC
#3 True CAVKDFNKFYF TGTGCCGTGAAAGACTTCAACAAATTTTACTTT
#4 True CASSLFSGTGTNEQFF TGTGCCAGCAGTTTATTTTCCGGGACAGGGACGAATGAGCAGTTCTTC
#5 True CAVKDFNKFYF TGTGCCGTGAAAGACTTCAACAAATTTTACTTT
#6 True CASSLFSGTGTNEQFF TGTGCCAGCAGTTTATTTTCCGGGACAGGGACGAATGAGCAGTTCTTC
# reads umis raw_consensus_id my.raw_clonotype_id clonotype.Freq
#1 1241 2 clonotype1_consensus_1 clonotype1 120
#2 2400 4 clonotype1_consensus_2 clonotype1 120
#3 1090 2 clonotype1_consensus_1 clonotype1 120
#4 2455 4 clonotype1_consensus_2 clonotype1 120
#5 1346 2 clonotype1_consensus_1 clonotype1 120
#6 3073 8 clonotype1_consensus_2 clonotype1 120
# proportion total.colonotype
#1 0.04098361 1292
#2 0.04098361 1292
#3 0.04098361 1292
#4 0.04098361 1292
#5 0.04098361 1292
#6 0.04098361 1292
# add it to iCellR object
my.obj <- add.vdj(my.obj, vdj.data = my.vdj.data)How to plot clonotypes
# once you have imported your clonotype data to your iCellR object, in order to plot them you need to have the following parapmeters:
# -1 clonotype name (e.g. clono = "clonotype1")
# -2 which column number has the clonotype names (e.g. clonotype.column = 2)
# -3 which column number has the cell barcode names (e.g. barcode.column = 1)
# In order to plot you need 2 things a- cell barcodes that match the barcodes in UMAP,PCA,tSNE or KNetL data and b- clonotype names.
# to check your clonotype data do this (example):
head(my.obj@vdj.data)
# raw_clonotype_id_SampleID MyBarcodes V1
#1 S5_clonotype98 Nor2.A_AAACCTGAGACAGACC.1 AAACCTGAGACAGACC.1
#2 S5_clonotype98 Nor2.A_AAACCTGAGACAGACC.1 AAACCTGAGACAGACC.1
#3 S4_clonotype100 Nor2.B_AAACCTGAGAGACTAT.1 AAACCTGAGAGACTAT.1
#4 S4_clonotype100 Nor2.B_AAACCTGAGAGACTAT.1 AAACCTGAGAGACTAT.1
#5 S3_clonotype3 Nor1.B_AAACCTGAGAGTCGGT.1 AAACCTGAGAGTCGGT.1
#6 S5_clonotype99 Nor2.A_AAACCTGAGATATGGT.1 AAACCTGAGATATGGT.1
# barcode SampleID raw_clonotype_id is_cell
#1 S5_AAACCTGAGACAGACC.1 5 clonotype98 True
#2 S5_AAACCTGAGACAGACC.1 5 clonotype98 True
#3 S4_AAACCTGAGAGACTAT.1 4 clonotype100 True
#4 S4_AAACCTGAGAGACTAT.1 4 clonotype100 True
#5 S3_AAACCTGAGAGTCGGT.1 3 clonotype3 True
#6 S5_AAACCTGAGATATGGT.1 5 clonotype99 True
# contig_id high_confidence length chain v_gene d_gene
#1 AAACCTGAGACAGACC-1_contig_2 True 514 TRB TRBV14 None
#2 AAACCTGAGACAGACC-1_contig_1 True 495 TRB TRBV20-1 None
#3 AAACCTGAGAGACTAT-1_contig_2 True 496 TRB TRBV9 None
#4 AAACCTGAGAGACTAT-1_contig_1 True 529 TRA TRAV26-1 None
#5 AAACCTGAGAGTCGGT-1_contig_1 True 512 TRB TRBV6-5 None
#6 AAACCTGAGATATGGT-1_contig_2 True 544 TRA TRAV12-2 None
# j_gene c_gene full_length productive cdr3
#1 TRBJ1-5 TRBC1 True True CASSFEGGSTQPQHF
#2 TRBJ2-7 TRBC2 True True CSARVRGRSSYEQYF
#3 TRBJ2-2 TRBC2 True True CASSVGVNTGELFF
#4 TRAJ52 TRAC True True CIVRGAGGTSYGKLTF
#5 TRBJ1-1 TRBC1 True True CASSYRPNTEAFF
#6 TRAJ33 TRAC True True CAVKRDSNYQLIW
# cdr3_nt reads umis
#1 TGTGCCAGCAGTTTTGAGGGGGGATCGACTCAGCCCCAGCATTTT 886 1
#2 TGCAGTGCTAGAGTAAGGGGACGGAGCTCCTACGAGCAGTACTTC 1912 3
#3 TGTGCCAGCAGCGTGGGCGTAAACACCGGGGAGCTGTTTTTT 10804 12
#4 TGCATCGTCAGGGGGGCTGGTGGTACTAGCTATGGAAAGCTGACATTT 960 4
#5 TGTGCCAGCAGTTACCGCCCGAACACTGAAGCTTTCTTT 4286 6
#6 TGTGCCGTGAAAAGGGATAGCAACTATCAGTTAATCTGG 1244 2
# raw_consensus_id my.raw_clonotype_id clonotype.Freq proportion
#1 clonotype98_consensus_1 S5_clonotype98 1 0.0001983930
#2 clonotype98_consensus_2 S5_clonotype98 1 0.0001983930
#3 clonotype100_consensus_2 S4_clonotype100 1 0.0001923817
#4 clonotype100_consensus_1 S4_clonotype100 1 0.0001923817
#5 clonotype3_consensus_1 S3_clonotype3 49 0.0070635721
#6 clonotype99_consensus_1 S5_clonotype99 1 0.0001983930
# total.colonotype
#1 5096
#2 5096
#3 5280
#4 5280
#5 5943
#6 5096
# In this example column number 1 and 2 have the clonotype and barcode info needed to plot.
# Sort clonotype names with highset frequency:
clonotype.frequency <- as.data.frame(sort(table(as.character(as.matrix((my.obj@vdj.data)[1]))),decreasing = TRUE))
head(clonotype.frequency)
# Var1 Freq
#1 S2_clonotype1 306
#2 S1_clonotype1 242
#3 S3_clonotype1 232
#4 S4_clonotype1 216
#5 S5_clonotype1 210
#6 S2_clonotype2 113
# let's plot S1_clonotype1 which is seen in 242 cells in all the conditions.
# if you want to plot only in one condtion or few conditions use this option "conds.to.plot" (e.g. conds.to.plot = c("WT","KO"))
# If conds.to.plot = NULL it would plot all of them (all 242 cells).
# Plot colonotype 1
clono.plot(my.obj, plot.data.type = "knetl",
clonotype.column = 1,
barcode.column = 2,
clono = "S1_clonotype1",
conds.to.plot = NULL,
cell.transparency = 1,
clust.dim = 2,
interactive = F)
# plot multiple clonotypes
ordered.clonotypes <- as.character(as.matrix((clonotype.frequency)[1]))
# let's plot top 19 clonotypes with highest frequency:
clonolist <- head(ordered.clonotypes, 19)
clonolist
# [1] "S2_clonotype1" "S1_clonotype1" "S3_clonotype1" "S4_clonotype1"
# [5] "S5_clonotype1" "S2_clonotype2" "S3_clonotype2" "S1_clonotype2"
# [9] "S2_clonotype4" "S1_clonotype4" "S3_clonotype4" "S2_clonotype3"
#[13] "S4_clonotype2" "S1_clonotype3" "S4_clonotype3" "S5_clonotype2"
#[17] "S3_clonotype3" "S2_clonotype9" "S3_clonotype6"
rm(list = ls(pattern="PL_"))
for(i in clonolist){
MyPlot <- clono.plot(my.obj, plot.data.type = "knetl",
clonotype.column = 1,
barcode.column = 2,
clono = i,
conds.to.plot = NULL,
cell.transparency = 1,
clust.dim = 2,
interactive = F)
NameCol=paste("PL",i,sep="_")
eval(call("<-", as.name(NameCol), MyPlot))
}
library(cowplot)
filenames <- ls(pattern="PL_")
B= cluster.plot(my.obj,plot.type = "knetl",interactive = F,cell.size = 0.5,cell.transparency = 1,anno.clust=TRUE)
filenames <- c("B",filenames)
png("19_clonotypes.png",width = 20, height = 20, units = 'in', res = 300)
plot_grid(plotlist=mget(filenames))
dev.off()

In this example the samples are normalized using DESeq2 so no normalization is needed.
sample.file.url = "https://genome.med.nyu.edu/results/external/iCellR/data/TCGA_sample_Normalized_data.tsv.gz"
download.file(url = sample.file.url,
destfile = "TCGA_sample_Normalized_data.tsv.gz",
method = "auto")
TCGA.data <- read.table("TCGA_sample_Normalized_data.tsv.gz")
head(TCGA.data)[1:3]
# Basal_TCGA.A1.A0SK.txt Basal_TCGA.A1.A0SP.txt Basal_TCGA.A2.A04P.txt
#TSPAN6 5823.4300 4318.034382 5265.733258
#TNMD 0.0000 6.049079 6.763079
#DPM1 3248.1536 2528.515113 1183.538813
#SCYL3 1059.7135 965.836315 1109.144945
#C1orf112 1251.3155 1070.687022 485.589067
#FGR 106.2438 933.574559 512.641383
library(iCellR)
my.obj <- make.obj(TCGA.data)
my.obj@main.data <- my.obj@raw.data
my.obj
###################################
,--. ,-----. ,--.,--.,------.
`--'' .--./ ,---. | || || .--. '
,--.| | | .-. :| || || '--'.'
| |' '--'\ --. | || || |
`--' `-----' `----'`--'`--'`--' '--'
###################################
An object of class iCellR version: 1.2.4
Raw/original data dimentions (rows,columns): 69797,882
Data conditions in raw data: Basal,Her2,LumA,LumB,Normal (131,64,404,170,113)
Row names: TSPAN6,TNMD,DPM1 ...
Columns names: Basal_TCGA.A1.A0SK.txt,Basal_TCGA.A1.A0SP.txt,Basal_TCGA.A2.A04P.txt ...
###################################
QC stats performed:FALSE, PCA performed:FALSE, CCA performed:FALSE
Clustering performed:FALSE, Number of clusters:0
tSNE performed:FALSE, UMAP performed:FALSE, DiffMap performed:FALSE
Main data dimentions (rows,columns):69797,882
Normalization factors:,...
Imputed data dimentions (rows,columns):0,0
############## scVDJ-Seq ###########
VDJ data dimentions (rows,columns):0,0
############## CITE-Seq ############
ADT raw data dimentions (rows,columns):0,0
ADT main data dimentions (rows,columns):0,0
ADT columns names:...
ADT row names:...
########### iCellR object ##########
my.obj <- run.pca(my.obj)
my.obj <- run.clustering(my.obj,
clust.method = "kmeans",
dist.method = "euclidean",
index.method = "silhouette",
max.clust =25,
min.clust = 2,
dims = 1:10)
my.obj <- run.pc.tsne(my.obj, dims = 1:10)
my.obj <- run.umap(my.obj, dims = 1:10, method = "umap-learn")
cluster.plot(my.obj,plot.type = "pca",cell.color = "black",col.by = "conditions",cell.transparency = 0.5,interactive = F)
cluster.plot(my.obj,plot.type = "umap",cell.color = "black",col.by = "conditions",cell.transparency = 0.5,interactive = F)
cluster.plot(my.obj,plot.type = "tsne",cell.color = "black",col.by = "conditions",cell.transparency = 0.5,interactive = F)
cluster.plot(my.obj,plot.type = "umap",cell.color = "black",cell.transparency = 1,interactive = F)

To do this you need to download the following databse files from our iCellR data link (more data to come soon).
# download the .rda files from here: https://genome.med.nyu.edu/results/external/iCellR/data/
# Load the .rda files as below
load("Immgen.GSE109125.205.rda")
load("Immgen.GSE122108.412.rda")
load("Immgen.GSE122597.83.rda")
load("Immgen.GSE124829.190.rda")
load("Immgen.microarray.GSE15907.653.rda")
load("Immgen.microarray.GSE37448.189.rda")
load("immgen.rna.rda")
load("immgen.uli.rna.rda")
load("mouse.cell.atlas.rda") | Key | Source | Samples | Description | Cell Types |
|---|---|---|---|---|
| GSE109125 | ImmGen | 205 | 83 populations representing all lineages and several differentiation cascades prepared from unchallenged mice and after LPS, anti-CD3, viral infection cell activation. | B Cells, Stromal Cells, Dendritic Cells, Granulocytes, Innate Lymphocytes, Stem Cells, Macrophages, ab T Cells, gd T Cells |
| GSE122108 | ImmGen | 412 | 130 populations comprising progenitors, residents, and stimulated (C.alb, LPS, injury, APAP+ starved overnight and pIC) mononuclear phagocytes for OpenSource MNP Project. | Macrophages, Kupffer Cell/Macrophages, Dendritic Cells, Microglia, Monocytes. |
| GSE122597 | ImmGen | 83 | Five highly purified immunocyte populations profiled to unusual depth as multiple replicates (8 to 16). Suitable for exploration of genes expressed at very low levels. | NK Cells, Follicular B, Naive CD4+ abT, gdT cells and peritoneal macrophages. |
| GSE124829 | ImmGen | 190 | 11 diverse immunocyte populations from male and female mice of varying ages stimulated with different dose of IFN to understand the immune system's sexual differences. | B Cells, Dendritic Cells, Neutrophils, Macrophages, Natural Killer T Cells, ab T Cells, gd T Cells, Microglia, Regulatory T Cells. |
| GSE15907 | ImmGen | 653 | 178 populations compromiing of gene-expression microarray datasets ("version1" labeling) from primary cells from multiple immune lineages are isolated ex-vivo, primarily from 6weeks B6 male mice. | gd T Cells, ab T Cells, Dendritic Cells, Macrophages, Stem Cells, B Cells, Stromal Cells, Neutrophils, Fibroblast, NK Cells, NK T Cells, Monocytes, CD4 Naive T Cell. |
| GSE37448 | ImmGen | 189 | 80 populations compromising of gene-expression microarray datasets ("version2" labeling) from primary cells from multiple immune lineages are isolated ex-vivo, primarily from 6weeks B6 male mice. Complements the V1 compendium with additional cells. Unfortunately, the version change in the labeling process, while more efficient, introduced some biases such that the two sections of the data can be compared grossly, but not at fine resolution (we tried...). | gd T Cells, ab T Cells, Dendritic Cells, Macrophages, Stem Cells, B Cells, Stromal Cells, Neutrophils, Fibroblast, NK Cells, NK T Cells, Monocytes, CD4 Naive T Cell. |
| rna | ImmGen | 23 | Full depth directional RNA sequencing was performed on the core ImmGen populations to generate reference datasets for the tissues from 5 week-old C57BL/6J (Jackson Laboratory) males and females, double-sorted by flow cytometry, per ImmGen cell preparation SOP. | B, CD4T, CD8T, DC, MQ,NK, NKT, Treg |
| uli.rna | ImmGen | 157 | ||
| mca | Mouse Cell Atlas | 43 tissues | Constructed as a basic scheme for the Mouse Cell Atlas using Microwell-seq. | Uterus, TrophoblastStemCells, Thymus, Testis, Stomach, Spleen, SmallIntestine, Prostate, Placenta, PeripheralBlood, Pancreas, Ovary, NeontalBrain, NeonatalSkin, NeonatalRib, NeonatalMuscle, NeonatalHeart, NeonatalCalvaria, Muscle, Mouse3T3, MesenchymalStemCellsPrimary, MesenchymalStemCells, MammaryGland.Virgin, MammaryGland.Pregnancy, MammaryGland.Lactation, MammaryGland.Involution, Male.fetal.Gonad, Lung, Liver, Kidney, FetalStomach, FetalLung, FetalLiver, FetalKidney, FetalIntestine, FetalBrain, Female.fetal.Gonad, EmbryonicStemCells, EmbryonicMesenchyme, Brain, BoneMarrowcKit, BoneMarrow, Bladder |
Choose a cluster and take for example top 10 genes for that cluster and then choose one of the databases that is best for you from the above list and predict your cell type. Note that if you have B cells for example and the database of your choice dose not have B cells, it would predict the closest looking cells to B cells. So it's important to use the right database for the right type of data.
# Choose top 40 genes for cluster 8 for example
MyGenes <- top.markers(marker.genes, topde = 40, min.base.mean = 0.2, cluster = 8)
####### predict
# plot
cell.type.pred(immgen.data = "rna", gene = MyGenes, plot.type = "point.plot")
cell.type.pred(immgen.data = "uli.rna", gene = MyGenes, plot.type = "point.plot", top.cell.types = 50)
cell.type.pred(immgen.data = "rna", gene = MyGenes, plot.type = "heatmap")
cell.type.pred(immgen.data = "uli.rna", gene = MyGenes, plot.type = "heatmap")
# As you can see cluster 8 is most likely to be B-cells.
# more examples
cell.type.pred(immgen.data = "GSE109125", gene = MyGenes, plot.type = "point.plot", top.cell.types = 50)
cell.type.pred(immgen.data = "GSE37448", gene = MyGenes, plot.type = "heatmap", top.cell.types = 50)
# for tissue type prediction use this:
cell.type.pred(immgen.data = "mca", gene = MyGenes, plot.type = "point.plot")
# And finally check the genes in the cells and find the common ones to predict
heatmap.gg.plot(my.obj, gene = MyGenes, interactive = F, cluster.by = "clusters")




You can automate this for all the clusters as below. Add as many plot as you wish.
Clusters = sort(unique(my.obj@best.clust$clusters))
for(i in Clusters){
Cluster = i
MyGenes <- top.markers(marker.genes, topde = 10, min.base.mean = 0.2, cluster = Cluster)
# first plot
Name <- paste("ImmGen_Cluster_",Cluster,"_pointPlot_RNA.pdf",sep="")
pdf(Name, width = 10, height = 10)
print(cell.type.pred(immgen.data = "rna", gene = MyGenes, plot.type = "point.plot"))
dev.off()
# second plot
Name <- paste("ImmGen_Cluster_",Cluster,"_check.pdf",sep="")
pdf(Name, width = 10, height = 10)
print(heatmap.gg.plot(my.obj, gene = MyGenes, interactive = F, cluster.by = "clusters"))
dev.off()
}- Pathway analysis
# Pathway
# pathways.kegg(my.obj, clust.num = 7)
# this function is being improved and soon will be available

In this example, we have downloaded 2 samples from 10X genomics website. You can get the data from these links: Anterior and Posterior. To make it easier you can also use the commands below to download from our server.
# download sample data
url = "https://genome.med.nyu.edu/results/external/iCellR/example7_Spatial_Transcriptomic/V1_Mouse_Brain_Sagittal_Anterior_Section_2_filtered_feature_bc_matrix.tar.gz"
# download the file
download.file(url = url,
destfile = "V1_Mouse_Brain_Sagittal_Anterior_Section_2_filtered_feature_bc_matrix.tar.gz",
method = "auto")
url ="https://genome.med.nyu.edu/results/external/iCellR/example7_Spatial_Transcriptomic/V1_Mouse_Brain_Sagittal_Anterior_Section_2_spatial.tar.gz"
# download the file
download.file(url = url,
destfile = "V1_Mouse_Brain_Sagittal_Anterior_Section_2_spatial.tar.gz",
method = "auto")
url ="https://genome.med.nyu.edu/results/external/iCellR/example7_Spatial_Transcriptomic/V1_Mouse_Brain_Sagittal_Posterior_Section_2_filtered_feature_bc_matrix.tar.gz"
# download the file
download.file(url = url,
destfile = "V1_Mouse_Brain_Sagittal_Posterior_Section_2_filtered_feature_bc_matrix.tar.gz",
method = "auto")
url ="https://genome.med.nyu.edu/results/external/iCellR/example7_Spatial_Transcriptomic/V1_Mouse_Brain_Sagittal_Posterior_Section_2_spatial.tar.gz"
# download the file
download.file(url = url,
destfile = "V1_Mouse_Brain_Sagittal_Posterior_Section_2_spatial.tar.gz",
method = "auto")
#########################
##### untar
untar("V1_Mouse_Brain_Sagittal_Anterior_Section_2_filtered_feature_bc_matrix.tar.gz")
untar("V1_Mouse_Brain_Sagittal_Anterior_Section_2_spatial.tar.gz")
file.rename("spatial","spatial_Anterior2")
file.rename("filtered_feature_bc_matrix","filtered_feature_bc_matrix_Anterior2")
untar("V1_Mouse_Brain_Sagittal_Posterior_Section_2_filtered_feature_bc_matrix.tar.gz")
untar("V1_Mouse_Brain_Sagittal_Posterior_Section_2_spatial.tar.gz")
file.rename("spatial","spatial_Posterior2")
file.rename("filtered_feature_bc_matrix","filtered_feature_bc_matrix_Posterior2")- Load the data
library(iCellR)
Anterior2 <- load10x("filtered_feature_bc_matrix_Anterior2",gene.name = 2)
Posterior2 <- load10x("filtered_feature_bc_matrix_Posterior2",gene.name = 2)
# if you want to analyze both samples
Samples <- c("Anterior2","Posterior2")
my.data <- data.aggregation(samples = Samples, condition.names = Samples)
# if you want to analyze 1 sample
# my.data <- load10x("filtered_feature_bc_matrix_Posterior2",gene.name = 2)
my.obj <- make.obj(my.data)
Anterior2 <- capture.image.10x("spatial_Anterior2")
Posterior2 <- capture.image.10x("spatial_Posterior2")
# if you want to analyze both samples
Samples <- c("Anterior2","Posterior2")
my.obj <- add.10x.image(my.obj,
image.data.list = Samples, condition.names = Samples)
# if one sample
# My.image <- image.capture.10x("Post2_spatial")
# my.obj <- add.10x.image(my.obj, image.data.list = "My.image")
my.obj
###################################
,--. ,-----. ,--.,--.,------.
`--'' .--./ ,---. | || || .--. '
,--.| | | .-. :| || || '--'.'
| |' '--'\ --. | || || |
`--' `-----' `----'`--'`--'`--' '--'
###################################
An object of class iCellR version: 1.6.0
Raw/original data dimentions (rows,columns): 31053,6118
Data conditions in raw data: Anterior2,Posterior2 (2825,3293)
Row names: A030001D20Rik,A030003K21Rik,A030005K14Rik ...
Columns names: Anterior2_AAACAAGTATCTCCCA.1,Anterior2_AAACACCAATAACTGC.1,Anterior2_AAACAGAGCGACTCCT.1 ...
###################################
QC stats performed:FALSE, PCA performed:FALSE
Clustering performed:FALSE, Number of clusters:0
tSNE performed:FALSE, UMAP performed:FALSE, DiffMap performed:FALSE
Main data dimensions (rows,columns): 0,0
Normalization factors:,...
Imputed data dimensions (rows,columns):0,0
############## scVDJ-seq ###########
VDJ data dimentions (rows,columns):0,0
############## CITE-seq ############
ADT raw data dimensions (rows,columns):0,0
ADT main data dimensions (rows,columns):0,0
ADT columns names:...
ADT row names:...
############## scATAC-seq ############
ATAC raw data dimensions (rows,columns):0,0
ATAC main data dimensions (rows,columns):0,0
ATAC columns names:...
ATAC row names:...
############## Spatial ###########
Spatial data dimentions (rows,columns):9984,5
########### iCellR object ##########The rest of the analysis is just like regular scRNA-Seq. Filter, normalize, run PCA, tSNE, UMAP, KNetL map and cluster. Then you can start ploting as below:
A=spatial.plot(my.obj,col.by = "clusters",conds.to.plot = "Anterior2",interactive= F)
B=spatial.plot(my.obj,col.by = "clusters",conds.to.plot = "Posterior2",interactive= F)
C= cluster.plot(my.obj,plot.type = "tsne",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
D= cluster.plot(my.obj,plot.type = "tsne",col.by = "conditions",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
E=spatial.plot(my.obj,col.by = "gene", gene = c("Cd4"), conds.to.plot = "Anterior2",interactive= F, scaleValue = TRUE)
F=spatial.plot(my.obj,col.by = "gene", gene = c("Cd4"), conds.to.plot = "Posterior2",interactive= F, scaleValue = TRUE)
library(gridExtra)
png('AllClusts.png', width = 8, height = 8, units = 'in', res = 300)
grid.arrange(A,B,C,D,E,F)
dev.off()

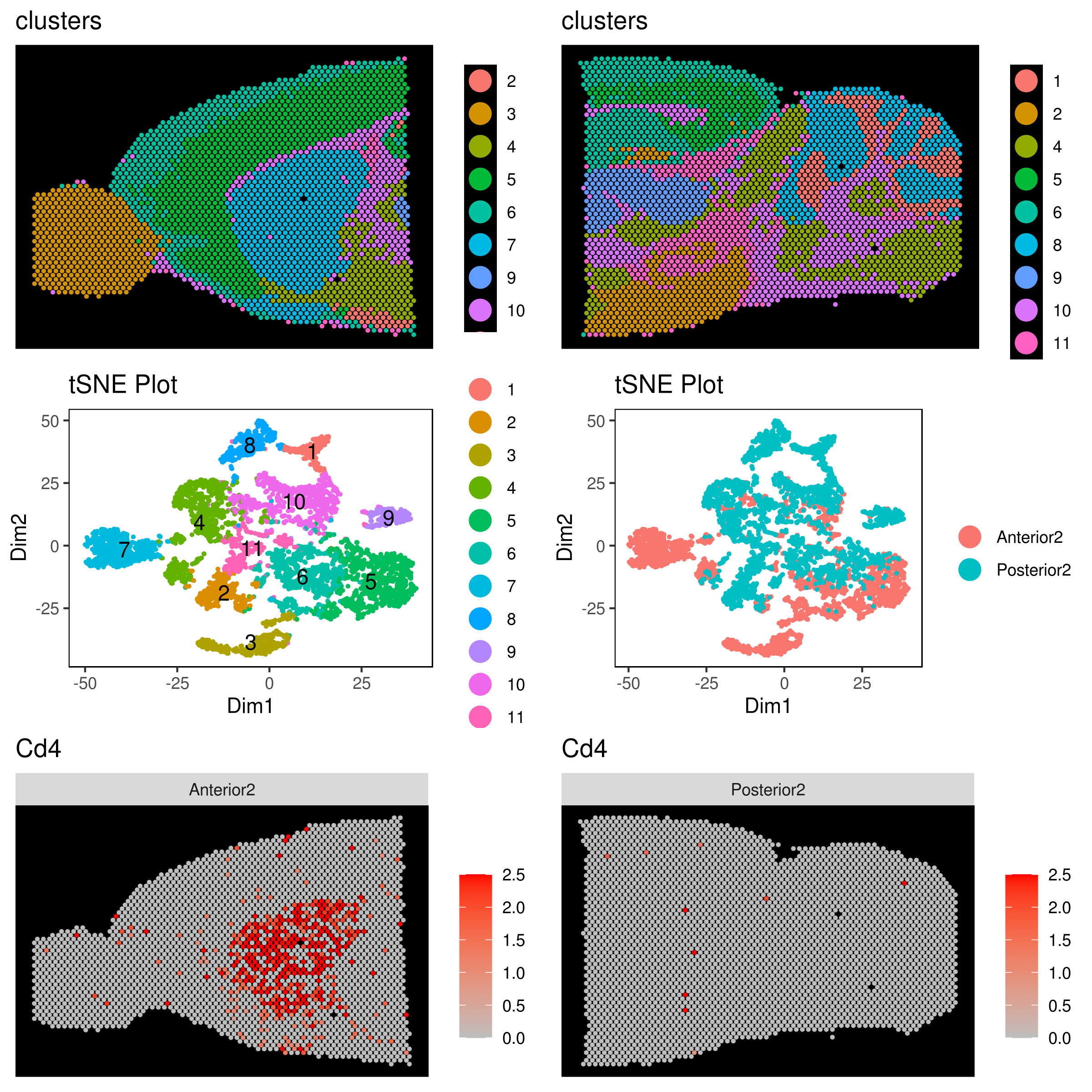
library("iCellR")
my.data <- load10x("filtered_gene_bc_matrices/")
# see the row names
row.names(my.data)
# get peak names
ATAC <- grep("^chr",row.names(my.data),value=T)
# get scATAC data
MyATAC <- subset(my.data, row.names(my.data) %in% ATAC)
head(MyATAC)[1:3]
# AAACAGCCAAGTGAAC.1 AAACAGCCACTGACCG.1 AAACAGCCATGATTGT.1
#chr1.181218.181695 0 0 1
#chr1.191296.191699 0 0 0
#chr1.629770.630129 0 0 0
#chr1.633806.634251 0 0 0
#chr1.778422.779040 0 0 0
#chr1.827306.827702 0 0 0
dim(MyATAC)
# [1] 21923 6326
# get RNA data
MyRNAs <- subset(my.data, !row.names(my.data) %in% ATAC)
head(MyRNAs)[1:3]
# AAACAGCCAAGTGAAC.1 AAACAGCCACTGACCG.1 AAACAGCCATGATTGT.1
#MIR1302.2HG 0 0 0
#FAM138A 0 0 0
#OR4F5 0 0 0
#AL627309.1 0 0 0
#AL627309.3 0 0 0
#AL627309.2 0 0 0
dim(MyRNAs)
#[1] 36633 6326
# make iCellR object
my.obj <- make.obj(MyRNAs)
# add ATAC-Seq data
my.obj@atac.raw <- MyATAC
my.obj@atac.main <- MyATAC
# check your object
my.obj
###################################
,--. ,-----. ,--.,--.,------.
`--'' .--./ ,---. | || || .--. '
,--.| | | .-. :| || || '--'.'
| |' '--'\ --. | || || |
`--' `-----' `----'`--'`--'`--' '--'
###################################
An object of class iCellR version: 1.6.2
Raw/original data dimentions (rows,columns): 24127,6326
Data conditions: no conditions/single sample
Row names: MIR1302.2HG,TTLL10.AS1,MRPL20.AS1 ...
Columns names: AAACAGCCAAGTGAAC.1,AAACAGCCACTGACCG.1,AAACAGCCATGATTGT.1 ...
###################################
QC stats performed:FALSE, PCA performed:FALSE
Clustering performed:FALSE, Number of clusters:0
tSNE performed:FALSE, UMAP performed:FALSE, DiffMap performed:FALSE
Main data dimensions (rows,columns): 0,0
Normalization factors:,...
Imputed data dimensions (rows,columns):0,0
############## scVDJ-seq ###########
VDJ data dimentions (rows,columns):0,0
############## CITE-seq ############
ADT raw data dimensions (rows,columns):0,0
ADT main data dimensions (rows,columns):0,0
ADT columns names:...
ADT row names:...
############## scATAC-seq ############
ATAC raw data dimensions (rows,columns):21923,6326
ATAC main data dimensions (rows,columns):21923,6326
ATAC columns names:AAACAGCCAAGTGAAC.1...
ATAC row names:chr1.181218.181695...
############## Spatial ###########
Spatial data dimentions (rows,columns):0,0
########### iCellR object ##########
From here do the regular scRNA-seq as expleind above. See example below
# QC
my.obj <- qc.stats(my.obj,
s.phase.genes = s.phase,
g2m.phase.genes = g2m.phase)
# plot as mentioned above
# filter
my.obj <- cell.filter(my.obj,
min.mito = 0,
max.mito = 0.07 ,
min.genes = 500,
max.genes = 4000,
min.umis = 0,
max.umis = Inf)
# normalize RNA
my.obj <- norm.data(my.obj, norm.method = "ranked.glsf", top.rank = 500)
# normalize ADT
my.obj <- norm.adt(my.obj)
# gene stats
my.obj <- gene.stats(my.obj, which.data = "main.data")
# find genes for PCA
my.obj <- make.gene.model(my.obj, my.out.put = "data",
dispersion.limit = 1.5,
base.mean.rank = 500,
no.mito.model = T,
mark.mito = T,
interactive = F,
no.cell.cycle = T,
out.name = "gene.model")
# run PCA and the rest is as above
my.obj <- run.pca(my.obj, method = "gene.model", gene.list = my.obj@gene.model,data.type = "main")
# tSNE
my.obj <- run.pc.tsne(my.obj, dims = 1:10)
# UMAP
my.obj <- run.umap(my.obj, dims = 1:10)
# KNetL
my.obj <- run.knetl(my.obj, dims = 1:20, zoom = 200, dim.redux = "umap")
# clustering based on KNetL
my.obj <- iclust(my.obj, sensitivity = 200, data.type = "knetl")
# clustering based on PCA
# my.obj <- iclust(my.obj, sensitivity = 100, data.type = "pca", dims=1:10)
# check clusters and adjust if needed (optinal)
# cluster.plot(my.obj,plot.type = "knetl",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
# my.obj <- change.clust(my.obj, change.clust = 3, to.clust = 4)
# my.obj <- change.clust(my.obj, change.clust = 3, to.clust = 10)
# order clusters
my.obj <- clust.ord(my.obj,top.rank = 500, how.to.order = "distance")
# plot
A= cluster.plot(my.obj,plot.type = "pca",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
B= cluster.plot(my.obj,plot.type = "umap",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
C= cluster.plot(my.obj,plot.type = "tsne",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
D= cluster.plot(my.obj,plot.type = "knetl",interactive = F,cell.size = 0.5,cell.transparency = 1, anno.clust=T)
library(gridExtra)
png('AllClusts.png', width = 12, height = 10, units = 'in', res = 300)
grid.arrange(A,B,C,D)
dev.off()
# save object
save(my.obj, file = "my.obj.Robj")
# find markers
marker.genes <- findMarkers(my.obj,
data.type = "main",
fold.change = 2,
padjval = 0.1,
uniq = F,
positive = T)
marker.genes1 <- cbind(row = rownames(marker.genes), marker.genes)
write.table((marker.genes1),file="marker.genes.tsv", sep="\t", row.names =F)
MyGenes <- top.markers(marker.genes, topde = 10, min.base.mean = 0.2, filt.ambig = F)
MyGenes <- unique(MyGenes)
png('heatmap_gg_genes.png', width = 10, height = 10, units = 'in', res = 300)
heatmap.gg.plot(my.obj, gene = MyGenes, interactive = F, cluster.by = "clusters",cell.sort = F, conds.to.plot = NULL)
dev.off()Work on scATAC data (normalize and find marker peaks for each cluster)
# normalize ACAT
my.obj <- norm.data(my.obj, norm.method = "ranked.glsf", top.rank = 500, ATAC.data = TRUE, ATAC.filter = TRUE)
marker.peaks <- findMarkers(my.obj,
data.type = "atac",
fold.change = 2,
padjval = 0.1,
uniq = F,
positive = T)
marker.peaks1 <- cbind(row = rownames(marker.peaks), marker.peaks)
write.table((marker.peaks1),file="marker.peaks.tsv", sep="\t", row.names =F)
head(marker.peaks1)
# row baseMean baseSD
#chr17.64986035.64986113 chr17.64986035.64986113 0.01217359 0.18257818
#chr1.26542287.26542678 chr1.26542287.26542678 0.05828764 0.80077656
#chr4.8199063.8199275 chr4.8199063.8199275 0.04280424 0.56205649
#chr20.50274929.50275237 chr20.50274929.50275237 0.04684509 0.63361490
#chr2.218382038.218382236 chr2.218382038.218382236 0.03122394 0.31153105
#chr11.1760469.1760814 chr11.1760469.1760814 0.07050175 0.63322284
# AvExpInCluster AvExpInOtherClusters foldChange
#chr17.64986035.64986113 0.07868394 0.002346603 33.530999
#chr1.26542287.26542678 0.33849093 0.016887273 20.044144
#chr4.8199063.8199275 0.24803497 0.012481148 19.872769
#chr20.50274929.50275237 0.27007513 0.013862584 19.482308
#chr2.218382038.218382236 0.17736269 0.009631770 18.414340
#chr11.1760469.1760814 0.39043782 0.023230813 16.806894
# log2FoldChange pval padj clusters
#chr17.64986035.64986113 5.067424 1.187697e-05 2.875415e-02 1
#chr1.26542287.26542678 4.325109 3.653916e-05 8.539202e-02 1
#chr4.8199063.8199275 4.312721 6.059691e-06 1.489472e-02 1
#chr20.50274929.50275237 4.284093 2.871301e-05 6.779143e-02 1
#chr2.218382038.218382236 4.202758 6.359572e-10 1.677019e-06 1
#chr11.1760469.1760814 4.070981 6.813447e-11 1.800794e-07 1
# gene cluster_1 cluster_2
#chr17.64986035.64986113 chr17.64986035.64986113 0.0786839378 0.000000000
#chr1.26542287.26542678 chr1.26542287.26542678 0.3384909326 0.008895062
#chr4.8199063.8199275 chr4.8199063.8199275 0.2480349741 0.038672840
#chr20.50274929.50275237 chr20.50274929.50275237 0.2700751295 0.028703704
#chr2.218382038.218382236 chr2.218382038.218382236 0.1773626943 0.000000000
#chr11.1760469.1760814 chr11.1760469.1760814 0.3904378238 0.004537037
# cluster_3 cluster_4 cluster_5 cluster_6
#chr17.64986035.64986113 0.000000000 0.0000000000 0.0006934750 0.0010038760
#chr1.26542287.26542678 0.031485714 0.0029244992 0.0052261002 0.0041264535
#chr4.8199063.8199275 0.000000000 0.0007226502 0.0027450683 0.0113212209
#chr20.50274929.50275237 0.027092857 0.0121741140 0.0041820941 0.0051516473
#chr2.218382038.218382236 0.004292857 0.0042095532 0.0006722307 0.0038561047
#chr11.1760469.1760814 0.071678571 0.0177288136 0.0141820941 0.0061099806
# cluster_7 cluster_8
#chr17.64986035.64986113 0.003099029 0.009080357
#chr1.26542287.26542678 0.047110680 0.044484375
#chr4.8199063.8199275 0.014819417 0.028651786
#chr20.50274929.50275237 0.025499029 0.028765625
#chr2.218382038.218382236 0.011988350 0.035508929
#chr11.1760469.1760814 0.042093204 0.050708705
MyGenes <- top.markers(marker.peaks, topde = 10, min.base.mean = 0.2, filt.ambig = F)
MyGenes <- unique(MyGenes)
png('heatmap_gg_peaks.png', width = 10, height = 10, units = 'in', res = 300)
heatmap.gg.plot(my.obj, gene = MyGenes, interactive = F, cluster.by = "clusters",cell.sort = F, conds.to.plot = NULL, data.type = "atac")
dev.off()
my.obj <- run.impute(my.obj,data.type = "knetl", nn = 10, ATAC.data = FALSE)
png('heatmap_gg_peaks.png', width = 10, height = 10, units = 'in', res = 300)
heatmap.gg.plot(my.obj, gene = MyGenes, interactive = F, cluster.by = "clusters",cell.sort = F, conds.to.plot = NULL, data.type = "atac.imputed")
dev.off()
## you can also find avarage peak intensity per cluster
my.obj <- clust.avg.exp(my.obj, data.type = "atac")
head(my.obj@clust.avg)
#gene cluster_1 cluster_2 ...
#chr1.100037799.100038931 0.38238731 0.36750000 ...
#chr1.100132733.100133298 0.11195725 1.13593827 ...
#chr1.100249637.100250160 0.09851425 0.09511728 ...
#chr1.100265992.100266479 0.06768394 0.17707407 ...
#chr1.10032488.10033387 0.35273705 0.14885802 ...
#chr1.100352150.100352921 0.12006088 0.00000000 ...
# find out which cluster has the highest number
dat <- as.data.frame(t((my.obj@clust.avg)[,-1]))
dat <- hto.anno(hto.data = dat)
head(dat$assignment.annotatio)
#[1] cluster_1 cluster_2 cluster_4 cluster_2 cluster_3 cluster_4
#8 Levels: cluster_1 cluster_2 cluster_3 cluster_4 cluster_5 ... cluster_8Peak analysis
# make a bed file per cluster from the marker.peaks file you made up here
make.bed(marker.peaks)
# load packages
library(ChIPseeker)
library(clusterProfiler)
# load genome
require(TxDb.Hsapiens.UCSC.hg38.knownGene)
txdb <- TxDb.Hsapiens.UCSC.hg38.knownGene
Anno="org.Hs.eg.db"
# load bed files
Mylist1 = list.files(pattern=".bed")
Mylist1
Mylist <- as.list(Mylist1)
NAMES <- gsub('_peaks.bed','',Mylist1)
names(Mylist) <- NAMES
files <- Mylist
files
# perform analysis (example)
promoter <- getPromoters(TxDb=txdb, upstream=3000, downstream=3000)
tagMatrixList <- lapply(files, getTagMatrix, windows=promoter)
pdf("Plot_ProfileLineAll.pdf")
plotAvgProf(tagMatrixList, xlim=c(-3000, 3000))
dev.off()
pdf('Plot_ProfileLine.pdf', width = 8, height = 10)
plotAvgProf(tagMatrixList, xlim=c(-3000, 3000), facet="row")
dev.off()
pdf("Plot_heatmaps.pdf", width = 50, height = 6)
tagHeatmap(tagMatrixList, xlim=c(-3000, 3000), color=NULL)
dev.off()
# annotate
peakAnnoList <- lapply(files, annotatePeak, TxDb=txdb,
tssRegion=c(-3000, 3000), verbose=FALSE)
# plot annotatin
pdf("Plot_AnnoBar.pdf")
plotAnnoBar(peakAnnoList)
dev.off()
############### peak annotation
peakAnnoList <- lapply(files, annotatePeak, TxDb=txdb,
tssRegion=c(-3000, 3000), verbose=FALSE, annoDb=Anno)
capture.output(peakAnnoList, file = "peakAnnoList.txt")
genes = lapply(peakAnnoList, function(i) as.data.frame(i))
lapply(1:length(genes), function(i) write.table(genes[[i]],
file = paste0(names(genes[i]), ".xls"),
row.names = FALSE, sep="\t"))
Merging scATAC files with different intervals (as dipicted in bedtools website)
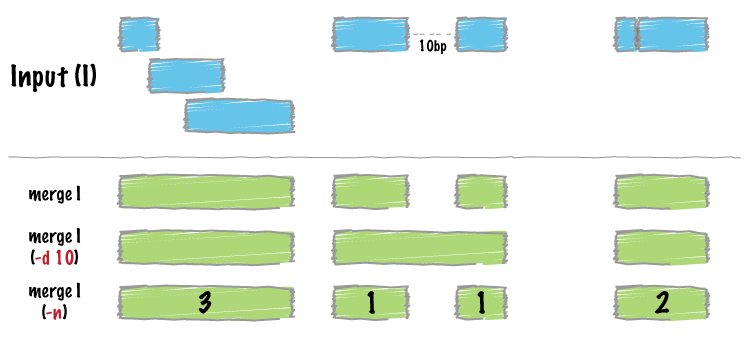
# Let's say you have 3 files that you need to merege
# example file
head(File1)[1:3]
# AAACAGCCAAGTGAAC.1 AAACAGCCACTGACCG.1 AAACAGCCATGATTGT.1
#chr1.181218.181695 0 0 1
#chr1.191296.191699 0 0 0
#chr1.629770.630129 0 0 0
#chr1.633806.634251 0 0 0
#chr1.778422.779040 0 0 0
#chr1.827306.827702 0 0 0
# get the row names from each file and concatenate them as below:
f1 <- row.names(File1)
f2 <- row.names(File2)
f3 <- row.names(File3)
all.peaks <- c(f1,f2,f3)
head(all.peaks)
#[1] "chr1.181218.181695" "chr1.191296.191699" "chr1.629770.630129"
#[4] "chr1.633806.634251" "chr1.778422.779040" "chr1.827306.827702"
chr <- as.character(as.matrix(data.frame(do.call('rbind', strsplit(as.character(all.peaks),'.',fixed=TRUE)))[1]))
start <- data.frame(do.call('rbind', strsplit(as.character(all.peaks),'.',fixed=TRUE)))[2]
end <- data.frame(do.call('rbind', strsplit(as.character(all.peaks),'.',fixed=TRUE)))[3]
# make a bed file
DAT <- as.data.frame(chr)
DAT$start <- as.numeric(as.matrix(start))
DAT$end <- as.numeric(as.matrix(end))
head(DAT)
# chr start end
#1 chr1 181218 181695
#2 chr1 191296 191699
#3 chr1 629770 630129
#4 chr1 633806 634251
#5 chr1 778422 779040
#6 chr1 827306 827702
# make a GenomicRanges object
library("GenomicRanges")
all.gr <- GRanges(seqnames=DAT$chr,ranges=IRanges(start=DAT$start,end=DAT$end))
all.gr
#GRanges object with ?? ranges and 0 metadata columns:
# seqnames ranges strand
# <Rle> <IRanges> <Rle>
# [1] chr1 181218-181695 *
# [2] chr1 191296-191699 *
# [3] chr1 629770-630129 *
# [4] chr1 633806-634251 *
# [5] chr1 778422-779040 *
# ... ... ... ...
# [52] chr1 1303892-1306216 *
# [53] chr1 1307242-1309359 *
# [54] chr1 1324425-1325236 *
# [55] chr1 1348940-1349958 *
# [56] chr1 1372031-1372220 *
# -------
# seqinfo: 1 sequence from an unspecified genome; no seqlengths
# sort and merge the peaks
mrg <- reduce(all.gr)
########################## choose file and give name
MyFile <- f1
name="f1_new.bed"
########################## copy paste the code here to make a new bed file
########################## the new bed has the old and new intervals (new intervals to be replaced with old)
chr <- as.character(as.matrix(data.frame(do.call('rbind', strsplit(as.character(MyFile),'.',fixed=TRUE)))[1]))
start <- data.frame(do.call('rbind', strsplit(as.character(MyFile),'.',fixed=TRUE)))[2]
end <- data.frame(do.call('rbind', strsplit(as.character(MyFile),'.',fixed=TRUE)))[3]
# make a bed file
DAT <- as.data.frame(chr)
DAT$start <- as.numeric(as.matrix(start))
DAT$end <- as.numeric(as.matrix(end))
MyFile <- DAT
# make intrval file to replace to new regions
MyFile.gr <- GRanges(seqnames=MyFile$chr,ranges=IRanges(start=MyFile$start,end=MyFile$end))
F <- subsetByOverlaps(MyFile.gr,mrg)
M <- subsetByOverlaps(mrg,MyFile.gr)
###
chr <- as.character(F@seqnames)
DAT <- as.data.frame(chr)
DAT$start <- F@ranges@start
DAT$end <- (F@ranges@start + F@ranges@width) - 1
DAT$new.chr<- as.character(M@seqnames)
DAT$new.start <- M@ranges@start
DAT$new.end <- (M@ranges@start + M@ranges@width) - 1
# diff
ADD <- setdiff(mrg,MyFile.gr)
L <- length(as.character(ADD@seqnames))
chr <-rep("NA",L)
DAT1 <- as.data.frame(chr)
DAT1$start <- rep("NA",L)
DAT1$end <- rep("NA",L)
DAT1$new.chr<- as.character(ADD@seqnames)
DAT1$new.start <- ADD@ranges@start
DAT1$new.end <- (ADD@ranges@start + ADD@ranges@width) - 1
Final.DAT <- rbind(DAT,DAT1)
##### Write
write.table(Final.DAT,name,row.names=FALSE,sep="\t", quote = FALSE)
# example file
# head(Final.DAT,10)
# chr start end new.chr new.start new.end
#1 chr1 181218 181695 chr1 181218 181695
#2 chr1 191296 191699 chr1 191296 191699
#3 chr1 629770 630129 chr1 629770 630129
#4 chr1 633806 634251 chr1 633806 634251
#5 chr1 778422 779040 chr1 778422 779040
#6 chr1 827306 827702 chr1 827306 827702
#7 NA NA NA chr1 904635 904943
#8 NA NA NA chr1 923684 924085
#9 NA NA NA chr1 940590 940783
#10 NA NA NA chr1 959052 959594
# The first 3 columns are the original peaks and the last 3 are the ones that need to be replaced with original one. The NA peaks would also get the new peak ids but in the matrix the cells will have 0 for expressions. To do this use the iCellR function replace.peak.id.
MyATAC1 <- replace.peak.id(atac.data=MyATAC1, bed.file = Final.DAT)
MyATAC2 <- replace.peak.id(atac.data=MyATAC2, bed.file = Final.DAT)
MyATAC3 <- replace.peak.id(atac.data=MyATAC3, bed.file = Final.DAT)
# finally aggregate the samples and add to iCellR object
my.atac.data <- data.aggregation(samples = c("MyATAC1","MyATAC2","MyATAC3"),
condition.names = c("WT","KO","Ctrl"))
# add ATAC-Seq data
[email protected] <- my.atac.data
[email protected] <- my.atac.data