fastqc_trimmomatic_normalization
Running FASTQC on the raw reads
% /usr/local/bin/fastqc wt37_rep1_1.fastq
You can see the files generated as output like so, which lists the files according to their time stamps, and newest files will show up at the bottom of the list:
% ls -ltr
.
...
-rw-r--r-- 1 brian brian 405767 May 31 20:25 wt37_rep1_1_fastqc.zip
-rw-r--r-- 1 brian brian 343555 May 31 20:25 wt37_rep1_1_fastqc.html
This generates an html file containing views into the quality of the reads. From your apache browser link, visit your workspace/ directory and view the above html file that was generated.
You should see a report like so:

In the event that quality trimming is required, we recommend using the Trimmomatic software. According to MacManes (2014), light trimming of reads at or below Phred=5 scores can improve upon the final transcriptome assembly. For example purposes, simply to demonstrate the effects of trimming on this high quality data set, we'll restrict it to the high value of 36, which should remove those initial bases with quality values < 36 and we should see the results of this in a subsequent run of fastqc on the trimmed reads.
Just to get some experience in running Trimmomatic, try executing it on our sample data as follows:
% java -jar $TRINITY_HOME/trinity-plugins/Trimmomatic/trimmomatic.jar \
PE -phred33 wt37_rep1_1.fastq wt37_rep1_2.fastq \
wt37_1.P.qtrim wt37_1.U.qtrim \
wt37_2.P.qtrim wt37_2.U.qtrim \
SLIDINGWINDOW:4:5 LEADING:36 TRAILING:36 MINLEN:25
.
TrimmomaticPE: Started with arguments: -phred33 wt37_rep1_1.fastq wt37_rep1_2.fastq wt37_1.P.qtrim wt37_1.U.qtrim wt37_2.P.qtrim wt37_2.U.qtrim SLIDINGWINDOW:4:5 LEADING:36 TRAILING:36 MINLEN:25
Multiple cores found: Using 16 threads
Input Read Pairs: 2000000 Both Surviving: 1879345 (93.97%) Forward Only Surviving: 94153 (4.71%) Reverse Only Surviving: 18098 (0.90%) Dropped: 8404 (0.42%)
TrimmomaticPE: Completed successfully
Running FASTQC on the quality-trimmed reads Normally, after quality trimming, you would confirm the improved read quality statistics by running FASTQC on the quality-trimmed reads. For example:
% /usr/local/bin/fastqc wt37_1.P.qtrim
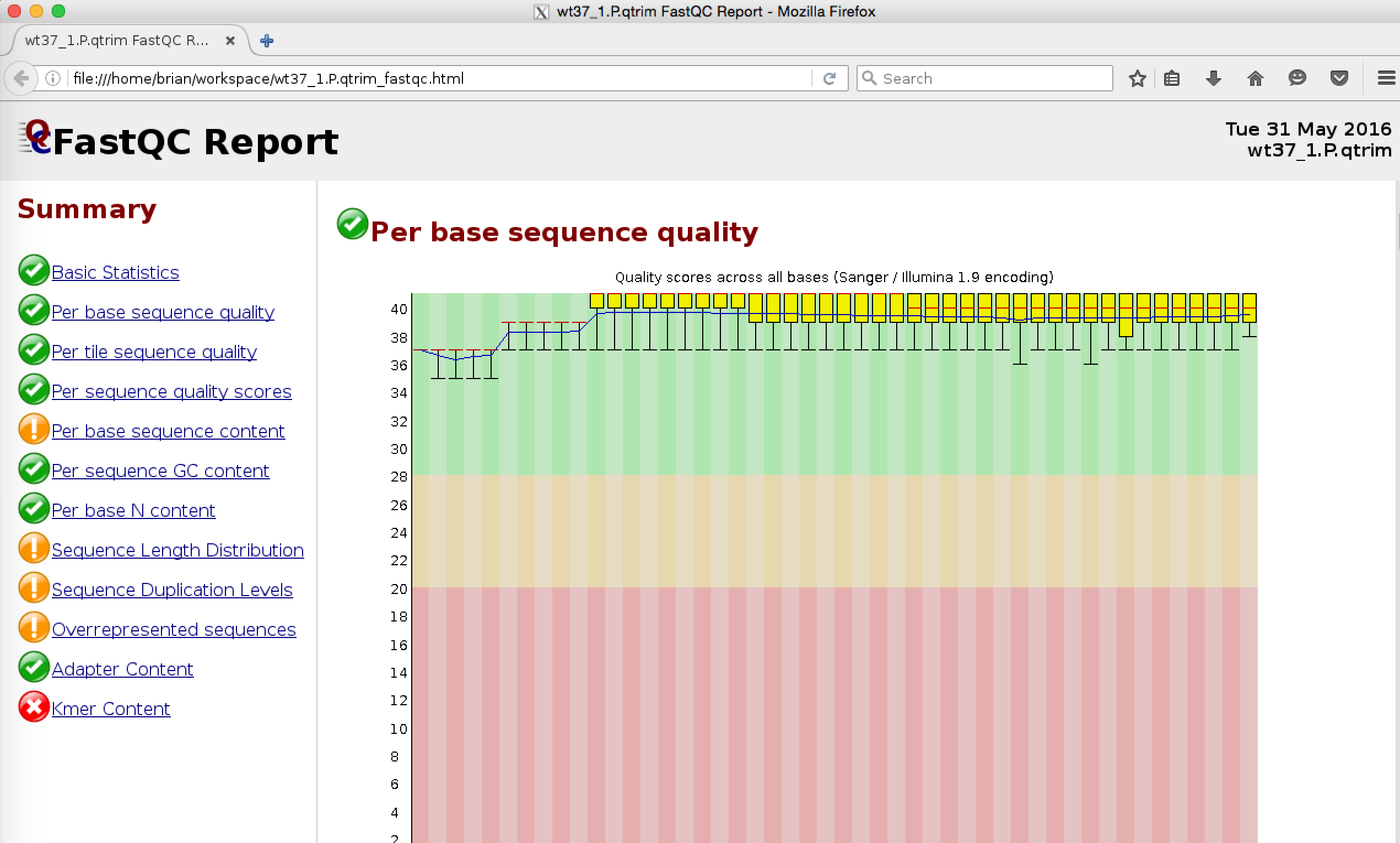
Note, in the context of running Trinity, it is not necessary that you manually run quality trimming on all of your reads. Instead, there are options available in Trinity to include automated trimmomatic trimming as shown above (Trinity parameter --trimmomatic).
Resources to examine:
- FASTQC website: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ and manual
- Trimmomatic website: http://www.usadellab.org/cms/?page=trimmomatic and manual.
- MacManes (2014) “On the optimal trimming of high-throughput mRNA sequence data. http://www.ncbi.nlm.nih.gov/pubmed/?term=24567737
Large rna-seq data sets will have a large excess of reads corresponding to moderately and highly expressed transcripts, and these are far more than what are needed for their assembly. By removing the excess reads, we can lower memory consumption and speed up the assembly process. In silico normalization is an effective way of identifying and removing those excess reads. (link to diginorm). We have a process integrated into Trinity to perform in silico normalization. Try running it like so, just for example purposes.
% $TRINITY_HOME/util/insilico_read_normalization.pl \
--seqType fq --JM 1G --max_cov 50 --left wt37_1.P.qtrim --right wt37_2.P.qtrim \
--pairs_together --output insil_norm_ex
.
1189570 / 1879312 = 63.30% reads selected during normalization.
1094 / 1879312 = 0.06% reads discarded as likely aberrant based on coverage profiles.
Normalization complete. See outputs:
/home/training/workspace/insil_norm_ex/wt37_1.P.qtrim.normalized_K25_C50_pctSD200.fq
/home/training/workspace/insil_norm_ex/wt37_2.P.qtrim.normalized_K25_C50_pctSD200.fq
Even with this small 2M set of paired reads, ~40% of the reads were removed as 'excess'. Note, this process only impacts the moderately and highly expressed transcripts. Any reads corresponding to lowly expressed transcripts should continue to exist in the normalized read sets.
Also note that normalization, like quality trimming, is - for the sake of convenience - integrated into Trinity itself, and normalization is on by default. To turn it off, you would use --no_normalize_reads with Trinity (but best to leave it on in most circumstances).