Dichotomous
Matt Wheeler Ph.D. 04/26/2023
This file shows ToxicR for dichotomous benchmark dose analyses using both single model and multiple model fits. The first thing we need to do is load the data. From an excel file this can be done using the following R commands:
dich_data <- matrix(0,nrow=5,ncol=3)
colnames(dich_data) <- c("Dose","N","Incidence")
dich_data[,1] <- c(0,50,100,200,400)
dich_data[,2] <- c(20,20,20,20,20)
dich_data[,3] <- c(0,1,2,10,19)Now suppose we want to fit a Weibull model to the data that is we have
the dose-response function
,
and want to estimate the probability of adverse response given the data.
How do we do this in ToxicR?
ToxicR has two main functions for fitting dichotomous dose-response models ‘single_dichotomous_fit’ and ma_dichotomous_fit.’ To fit any individual model, we use the same interface and use ‘single_dichotomous_fit’. This function takes several parameters, but to start, all we will do is specify the necessary ones. Here, the first argument is a vector of doses, the second is a vector of observed adverse events, and the third is a vector containing the number of animals in a given dose group.
library(ToxicR)##
##
## _______ _____
## |__ __| 🤓 | __ \
## | | _____ ___ ___| |__) |
## | |/ _ \ \/ / |/ __| _ /
## | | (_) > <| | (__| | \ \
## |_|\___/_/\_\_|\___|_| \_\
## 23.4.1.1.0
## ___
## | |
## / \ ____()()
## /☠☠☠\ / xx
## (_____) `~~~~~\_;m__m.__>o
##
##
## THE SOFTWARE IS PROVIDED AS IS, WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED,
## INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A
## PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT
## HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF
## CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE
## OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.
library(ggplot2)
weibull_fit <- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="weibull")The previous line fits the Weibull Bayesian Laplace model and puts all of the information one needs in the `weibull_fit’ object variable. Say we want to get the BMD and associated 95% confidence interval on the BMD. All we need to do is look in the object.
weibull_fit$bmd## BMD BMDL BMDU
## 86.77428 54.42931 125.40872
Here, we see the BMD is estimated to be
with a
BMDL of
which matches the Bayesian Laplace fits in the US-EPA’s software. The
`weibull_fit’ object contains all of the information you are used to
and some information not in the standard BMDS analysis. Here are just
three different options.
# Model Parameters
weibull_fit$parameters## [1] -3.477066e+00 2.069425e+00 1.026415e-05
# Covariance estimate on those parameters
weibull_fit$covariance## [,1] [,2] [,3]
## [1,] 0.7958299464 0.09158446 -0.0006105861
## [2,] 0.0915844581 0.17517901 0.1903619359
## [3,] -0.0006105861 0.19036194 0.5206126021
# The prior used
weibull_fit$prior## Weibull Model [binomial] Parameter Priors
## ------------------------------------------------------------------------
## Prior [logit(g)]:Normal(mu = 0.00, sd = 2.000) 1[-20.00,20.00]
## Prior [a]:Log-Normal(log-mu = 0.42, log-sd = 0.500) 1[0.00,40.00]
## Prior [b]:Log-Normal(log-mu = 0.00, log-sd = 1.500) 1[0.00,10000.00]
The prior information also gives you the information on the parameter
interpretation. Notice that the
parameter is parameterized using the logit transform,
i.e.
which implies
To get the value as the background probability, we can run the following
code.
# Model Parameters
logit_g = weibull_fit$parameters[1]
1/(1+exp(-logit_g))## [1] 0.02997187
ToxicR is equipped with a powerful graphics engine using
ggplot2' graphics, and to print any fit, all one needs to do is typeplot(
xxx )’, where xxx is the variable name. This command will return a
ggplot object that can either be plotted immediately or customized with
further ggplot commands.
library(ggplot2) # This is needed to make changes
# Plot immediately
plot(weibull_fit)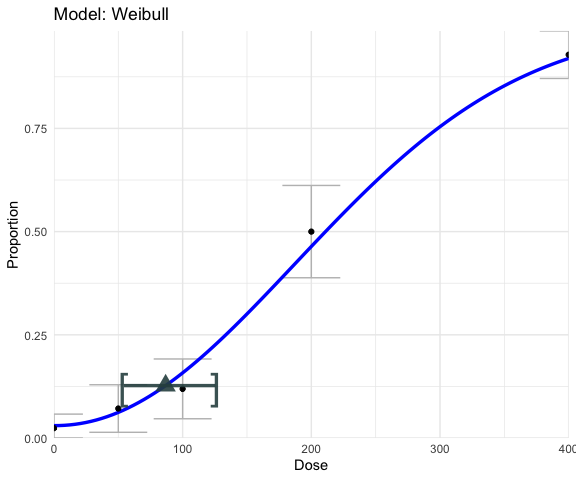
# Save fit and modify
ggplot_fit <- plot(weibull_fit)
# Transform the x axis
ggplot_fit + scale_x_continuous(trans = "pseudo_log")## Scale for x is already present.
## Adding another scale for x, which will replace the existing scale.
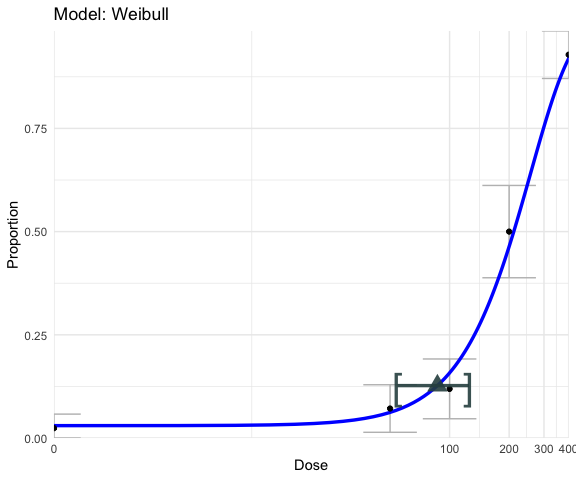
# Change labels
ggplot_fit + xlab("log(Dose) mg/kg/day") + ylab("Tumor Probability") +
scale_x_continuous(trans = "pseudo_log") + ggtitle("Weibull Fit") + theme_classic()## Scale for x is already present.
## Adding another scale for x, which will replace the existing scale.

We do not have time to go over all ‘ggplot’ commands, but the figure can be modified to your specifications.
In the above, we used the Bayesian Maximum A-Posteriori Laplace method, which is the default if you do not specify the type of fit. ToxicR allows for Laplace MAP, Maximum Likelihood, and MCMC fitting. Below, we fit all three options and then compare them in one plot.
# Laplace Explicitly Specified
weibull_fit_MAP <- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="weibull",fit_type="laplace")
# Maximum Likelihood Estimation
weibull_fit_MLE <- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="weibull",fit_type="mle")
# MCMC Estimation
weibull_fit_MCMC <- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="weibull",fit_type="mcmc")
library(ggpubr)
figure <- ggarrange(plot(weibull_fit_MAP)+ggtitle(""), plot(weibull_fit_MLE)+ggtitle(""),
plot(weibull_fit_MCMC)+ggtitle(""),
labels = c("MAP", "MLE", "MCMC"),
ncol = 2, nrow = 2)
figure
Here, the fits are essentially the same. This is not always the case.
Notice how the mcmc fit has additional graphics. These additions are
because of the nature of MCMC sampling. Here, we add the uncertainty in
the curve estimation and BMD distribution information. This plot shows
bounds and the estimated posterior BMD distribution. For dichotomous
models, there are three additional things you can specify. The most
important of which is the benchmark response (BMR). By default it is set
to
but it can be any value between zero and 1, i.e.,
# Laplace Explicitly Specified
weibull_fit_MCMC_1 <- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="weibull",fit_type="mcmc",samples = 75000)
# Maximum Likelihood Estimation
weibull_fit_MCMC_2<- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="weibull",fit_type="mcmc", BMR=0.05,samples = 7500)
# MCMC Estimation
weibull_fit_MCMC_3 <- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="weibull",fit_type="mcmc", BMR =0.01,samples = 7500)
fig_1 <- plot(weibull_fit_MCMC_1) + ggtitle("") + scale_x_continuous(trans="pseudo_log")## Scale for x is already present.
## Adding another scale for x, which will replace the existing scale.
fig_2 <- plot(weibull_fit_MCMC_2) + ggtitle("") + scale_x_continuous(trans="pseudo_log")## Scale for x is already present.
## Adding another scale for x, which will replace the existing scale.
fig_3 <- plot(weibull_fit_MCMC_3) + ggtitle("") + scale_x_continuous(trans="pseudo_log")## Scale for x is already present.
## Adding another scale for x, which will replace the existing scale.
figure <- ggarrange(fig_1, fig_2, fig_3,
labels = c("BMR=0.1", "BMR=0.05", "BMR=0.01"),
ncol = 2, nrow = 2)
figure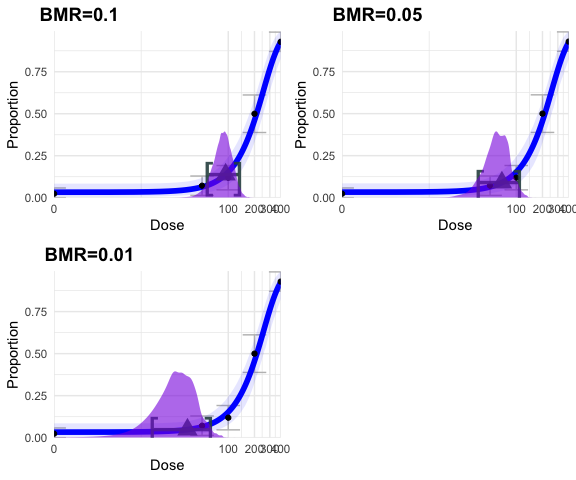
In the above code, notice `samples = 75000,’ which specifies
MCMC samples instead of the default
Of the remaining options, the last one allows the user to change the
default prior. Here, we give the user freedom, but this is given with
the warning: “USE AT YOUR OWN RISK!” To specify a prior, you use several
functions ‘normprior,’ ‘lnormprior,’ ‘create_prior list,’ and
‘create_dichotomous_prior.’ The functions ‘normprior’ and ‘lnormprior’
define the individual parameter priors to be normal or log-normal,
respectively. The first argument is the mean/log-mean and the second
function argument is the standard deviation / log-standard deviation.
The final two give the parameter bounds, e.g., the lower and upper
bounds. If you do not want the parameter to be bounded, we recommend
placing -1e6 and 1e6 on the parameter. For estimation, this is
essentially numerically unbounded.
# Laplace Explicitly Specified
prior <- create_prior_list(normprior(0,10,-100,100),
lnormprior(0,1,0,100),
lnormprior(0,1,0,100));
PR1 <- create_dichotomous_prior(prior,"weibull")
prior <- create_prior_list(normprior(0,10,-100,100),
lnormprior(0,0.1,0,100),
lnormprior(0,0.1,0,100));
PR2 <- create_dichotomous_prior(prior,"weibull")
PR1## Weibull Model [binomial] Parameter Priors
## ------------------------------------------------------------------------
## Prior [logit(g)]:Normal(mu = 0.00, sd = 10.000) 1[-100.00,100.00]
## Prior [a]:Log-Normal(log-mu = 0.00, log-sd = 1.000) 1[0.00,100.00]
## Prior [b]:Log-Normal(log-mu = 0.00, log-sd = 1.000) 1[0.00,100.00]
PR2 ## Weibull Model [binomial] Parameter Priors
## ------------------------------------------------------------------------
## Prior [logit(g)]:Normal(mu = 0.00, sd = 10.000) 1[-100.00,100.00]
## Prior [a]:Log-Normal(log-mu = 0.00, log-sd = 0.100) 1[0.00,100.00]
## Prior [b]:Log-Normal(log-mu = 0.00, log-sd = 0.100) 1[0.00,100.00]
weibull_fit_prior1 <- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="weibull",fit_type="mcmc",
BMR=0.05,samples = 75000,prior=PR1)
weibull_fit_prior2 <- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="weibull",fit_type="mcmc",
BMR=0.05,samples = 75000,prior=PR2)We can then compare the two fits. Here, it is seen how the tight prior
on
constrains the fit and causes an esitmate very different than the
maximum likelihood estimate. This second estimate is very similar to the
quantal linear model
library(ggpubr)
figure <- ggarrange(plot(weibull_fit_prior1)+ggtitle(""),
plot(weibull_fit_prior2)+ggtitle(""),
plot(weibull_fit_MLE)+ggtitle(""),
labels = c("Prior-1", "Prior-2","MLE"),
ncol = 2, nrow = 2)
figure 
Given that one can change priors and look at the MLE estimate simultaneously, we can analyze prior choice in relation to BMD estimates. Here, we stress that the MLE or a diffuse prior is not always ‘the most objective .’ We are currently finishing up research on prior development to guide researchers and practitioners alike.
library(ggpubr)
#Compare the BMDs
rbind(weibull_fit_prior1$bmd,weibull_fit_prior2$bmd)## BMD BMDL BMDU
## [1,] 60.06219 33.48858 89.44485
## [2,] 22.76929 13.88375 33.40649
#Extract the BMD CDF and remove possible infinities
#that may occur because of asymptotes etc.
#Note ecdf() is the emperical CDF function in R
temp <- ecdf(weibull_fit_prior1$mcmc_result$BMD_samples)
plot(temp,col=1,main="BMD prior comparison",xlab="BMD mg/kg/day",xlim=c(0,200),lwd=2)
temp <- ecdf(weibull_fit_prior2$mcmc_result$BMD_samples)
lines(temp,col=2,lwd=2)
## The default MCMC fit
temp <- ecdf(weibull_fit_MCMC$mcmc_result$BMD_samples)
lines(temp,col=3,lwd=2)
#For the MLE comparison
#The information is in the MLE/MAP objects object, though it is
# a little different.
library(tidyverse)## ── Attaching packages ─────────────────────────────────────── tidyverse 1.3.2 ──
## ✔ tibble 3.2.1 ✔ dplyr 1.1.2
## ✔ tidyr 1.3.0 ✔ stringr 1.5.0
## ✔ readr 2.1.2 ✔ forcats 1.0.0
## ✔ purrr 1.0.1
## ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
## ✖ dplyr::filter() masks stats::filter()
## ✖ dplyr::lag() masks stats::lag()
temp <- as.data.frame(weibull_fit_MLE$bmd_dist)
names(temp) <- c("BMD","PROB")
temp <- temp %>% filter(!is.infinite(BMD))
lines(temp$BMD,temp$PROB,col=4,lwd=2)
In the last plot, we see the impacts different prior information has on
the BMD distributions. The red line is the most conservative prior, the
green line is the default prior, and the blue line represents the
approximate BMD distribution produced by MLE. Note how similar the blue
and green lines are. In general, the default priors will produce
behavior similar to the MLE when the data support estimation of all
parameters. The estimates will diverge when specific parameters are
weakly informed by the data, e.g.,
in the Weibull model.
Everything above can be done with a suite of models, and is callable using the function call ‘single_dichotomous_fit.’ To change the model, simply change the model_type. The following models are possible in ToxicR
and these models can be fit by setting the ‘model_type’ parameter to
‘weibull’,‘gamma’,‘logistic’,
‘log-logistic’,‘probit’,‘log-probit’,‘multistage’,‘qlinear’, and ‘hill’
respectively. For the multistage model the degree parameter, specified
as
above, defaults to 2, but can be modified by specifying the ‘degree’ in
the function call. For example, if one wants a 3 degree multistage
model, one would specify ‘degree = 3’. Finally the background parameter
is parameterized as
which implies if you want the probability it represents you need to
transform it as
The following code runs the same data using different models using Bayesian MAP-Laplace methods and then them.
qlinear_fit <- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="qlinear" )
gamma_fit <- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="gamma" )
hill_fit <- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="hill" )
lprobit_fit <- single_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],model_type="log-probit" )
library(ggpubr)
figure <- ggarrange(plot(qlinear_fit)+ggtitle(""),
plot(gamma_fit)+ggtitle(""),
plot(hill_fit)+ggtitle(""),
plot(lprobit_fit)+ggtitle(""),
labels = c("Quantal Linear", "Gamma","Hill","Log-Probit"),
ncol = 2, nrow = 2)
figure
Now this is the ‘old way.’ The new way is model averaging. Here, Toxic R supports this framework. Model averaging is much like the above framework, and by default, will use all nine models, their default priors, and Bayesian MAP-Laplace estimation (the other option is ‘mcmc’). For MA estimation, we use multiple CPU cores (except on MacOS). Thus, if one has 8+ cores, MA will take about the same time as a single model fit.
ma_fit <- ma_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"] )
plot(ma_fit)
ma_fit_mcmc <- ma_dichotomous_fit(dich_data[,"Dose"],dich_data[,"Incidence"],
dich_data[,"N"],fit_type='mcmc' )
plot(ma_fit_mcmc)
In addition to the standard plots we give additional plots, so one can compare BMDs. For MA-Laplace, we can call a ‘cleveland_plot()’. For MCMC, one can call ‘MAdensity_plot().’
library(ggplot2)
cleveland_plot(ma_fit)+ggtitle("Cleveland Plot MA-Laplace")## Warning: Removed 1 rows containing missing values (`geom_vline()`).

MAdensity_plot(ma_fit_mcmc)+ggtitle("Density Plot MCMC")## Picking joint bandwidth of 2.31

