2. RNA Seq Workflow_Introduction
RNA seq is widely used for gene expression studies to quantify the RNA in a sample using next-generation sequencing (NGS). It is a powerful tool with many applications for gene discovery and quantification. Here, we assume differential expression is being assessed between 2 experimental conditions, i.e. a simple 1:1 comparison. The sample data is from a human genome. It is highly reccomended to use a HPC environment for increased RAM and computational power.
The pipeline for this mini-project include:
-
FastQCfor quality check -
Trimmomaticfor adaptor removal and trimming -
HISAT2for alignment and Subread’sfeatureCountsfor count generation -
Kallistofor pseudoalignment -
MultiQCto collect the statistics
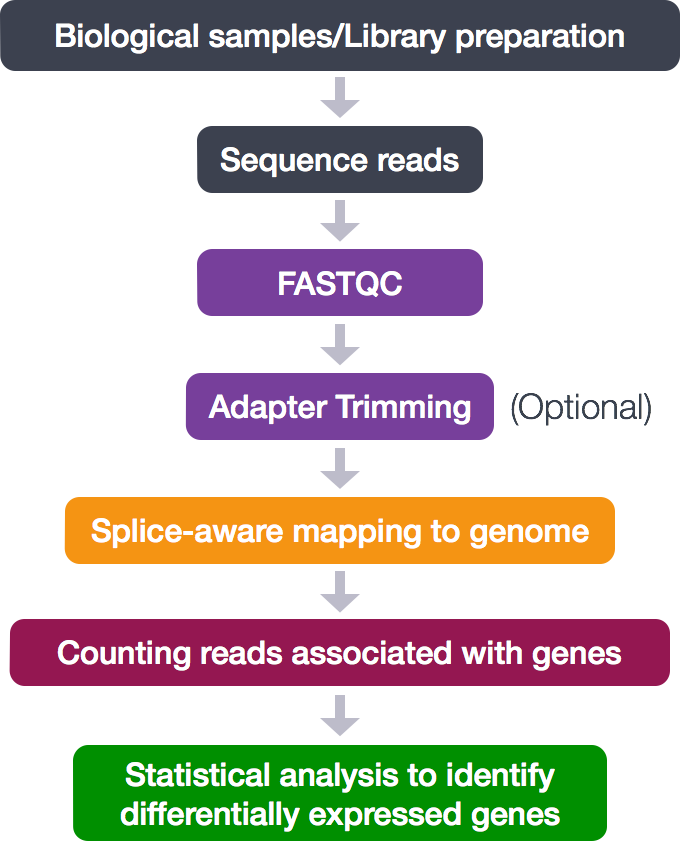
- Statistical analysis in R using
DESEQ - Converting the pipeline to R Markdown
- Converting the pipeline to a Snakemake pipeline
-
Download Miniconda for your specific OS to your home directory
- Linux:
wget https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh - Mac:
curl https://repo.continuum.io/miniconda/Miniconda3-latest-MacOSX-x86_64.sh
- Linux:
- Run:
bash Miniconda3-latest-Linux-x86_64.shbash Miniconda3-latest-MacOSX-x86_64.sh
- Follow all the prompts: if unsure, accept defaults
- Close and re-open your terminal
- If the installation is successful, you should see a list of installed packages with
-
conda listIf the command cannot be found, you can add Anaconda bin to the path using:export PATH=~/miniconda3/bin:$PATH
-
Clone this repository with folder structure into the current working folder
git clone https://github.com/nanjalaruth/Group-5-miniproject_RNASEQ.git
mkdir -p Rawdata Results Scripts README.mdcd Rawdata/
#nano sample_id.txt and then add a list of the sample names:
#sample37
#sample38
#sample39
#sample40
#sample41
#sample42
for sample in `cat sample_id.txt`
do
wget http://h3data.cbio.uct.ac.za/assessments/RNASeq/practice/dataset/${sample}_R1.fastq.gz
wget http://h3data.cbio.uct.ac.za/assessments/RNASeq/practice/dataset/${sample}_R2.fastq.gz
done
#download the metadata file
wget -c http://h3data.cbio.uct.ac.za/assessments/RNASeq/practice/practice.dataset.metadata.tsv